AI-Based Structure Prediction & Analysis
No protein structure means no rational drug design — yet most discovery teams wait months for crystallography. We deliver production-grade structural coordinates, quality assessments, and druggability insights — from AI-driven prediction to Cryo-EM/X-ray validation — under one roof.
Creative Biostructure at a Glance
Why Partner With Us
Most structure determination programs fail not because the target is intractable — but because the workflow is fragmented. Virtual biotechs burn runway coordinating a bioinformatics contractor, a protein expression shop, and a crystallography facility. Pharma teams lose months to handoff delays between computational predictions and experimental validation. We built Creative Biostructure to eliminate that friction: one platform where AI-driven prediction, experimental data processing, and structural quality assessment share the same project team, the same data system, and the same milestone clock.
Your CapEx is in compute and chemistry. Ours is in structural biology infrastructure.
| Stage | What We Deliver | What You Don't Need to Build |
|---|---|---|
| Sequence-to-Structure | AlphaFold2/RoseTTAFold prediction, homology modeling, ab initio folding | GPU cluster, ML engineering team |
| Experimental Data Processing | X-ray diffraction data analysis, Cryo-EM/ET image reconstruction, NMR spectral assignment | Synchrotron beamline, EM facility, NMR spectrometer |
| Structure Validation & Refinement | Model quality assessment, structure refinement, PDB deposition support | Crystallography software licenses, structural bioinformatics team |
| Structure-to-Insight | Binding pocket druggability analysis, protein-protein complex prediction, antibody structure prediction | CADD infrastructure |
Production-Ready Deliverables: Every structure ships with confidence scores (pLDDT/pTM), experimental validation data (as applicable), binding pocket analysis, and docking-ready coordinates — your medicinal chemists can enter virtual screening immediately, or our Hit Identification team can advance the program.
- ✓ Milestone-based pricing aligned with your fundraising cycles
- ✓ No vendor coordination overhead — prediction, data processing, and validation under a single project manager
Membrane proteins. Large complexes. Nucleic acid-protein assemblies. Antibody-antigen interfaces.
"Undruggable" is our starting point.
Proven track record where others fail
GPCRs, ion channels, transporters, nucleic acid-protein complexes, and PPI interfaces — targets that crash standard prediction pipelines due to lack of templates or multi-domain architecture.
Multi-modal pivot capability
When AlphaFold2 yields low-confidence regions, we deploy Cryo-EM data processing for experimental validation; when experimental phasing is stuck, AI-assisted molecular replacement breaks the deadlock.
IP firewall & encrypted data infrastructure
Full audit trails, GLP-ready documentation, client retains 100% ownership of all structural data and model parameters.
Core Service Modules
Service Module At-a-Glance
| Service | Core Capability | Structural + Computational Integration | Typical Timeline |
|---|---|---|---|
| AlphaFold Protein Structure Prediction | End-to-end AlphaFold2/RoseTTAFold pipeline; MSA optimization; custom template integration | Molecular docking and MD simulations for model validation; pocket druggability scoring | 1–2 weeks |
| Homology Modeling & Threading | Template identification (BLAST/HHpred); loop modeling; side-chain optimization; model quality scoring | Structure-based virtual screening against modeled pockets; pharmacophore modeling for ligand-guided refinement | 2–4 weeks |
| Ab Initio Modeling & Co-evolutionary Analysis | Contact prediction from MSA (GREMLIN/CCMPred); fragment assembly; deep learning folding for orphan targets | Binding pocket prediction on de novo folds; docking validation of predicted binding sites | 3–6 weeks |
| Structure Refinement & Quality Assessment | MolProbity/PROCHECK validation; Ramachandran analysis; B-factor refinement; EMRinger scoring for Cryo-EM | FEP calculations and MD stability tests to validate structural integrity before downstream design | 1–2 weeks |
| Structural Data Processing Services | X-ray diffraction data processing (HKL2000/XDS → MR → refinement); Cryo-EM/ET SPA (RELION/cryoSPARC) | AI-assisted model building (Phenix/DeepTracer); docking into experimental density maps | 4–12 weeks |
| Binding Pocket & Druggability Analysis | Pocket detection (FPocket/Concavity); druggability scoring (DrugScore/FTMap); allosteric site identification | Virtual screening and pharmacophore screening against prioritized pockets; ADMET pre-filtering | 2–3 weeks |
| Complex Structure Prediction | Protein-protein docking (HADDOCK/ClusPro); peptide-protein interaction modeling; multi-component assembly prediction | MD simulations for complex stability; binding free energy calculations (MM/PBSA) for affinity ranking | 3–6 weeks |
| Antibody Structure Prediction | IgFold/DeepAb for Fv/VH-VL orientation; CDR loop refinement; antibody-antigen docking | Affinity maturation simulation; epitope mapping; humanization scoring | 1–3 weeks |
AlphaFold Protein Structure Prediction
Production-Grade AI Folding with Experimental Validation
Key Features:
- Optimized MSA Construction — Custom sequence database curation and MSA depth optimization for eukaryotic, membrane-bound, and multi-domain targets.
- Template-Enhanced Folding — Integration of experimental structures, homology models, or Cryo-EM maps as folding templates to boost confidence in critical regions.
- Confidence-Gated Downstream Design — pLDDT/pTM scores directly inform virtual screening strategy: high-confidence pockets enter SBVS; low-confidence regions trigger FBDD or experimental Cryo-EM validation.
What We Offer: For virtual biotechs, structural starting points within days of receiving a FASTA file — no protein expression delay. For pharma, high-throughput structural coverage of entire target portfolios, each model validated against experimental data and flagged for risk where confidence is low.
Explore AlphaFold Prediction →Homology Modeling & Threading
Template-Based Modeling for Targets with Known Folds

Key Features:
- Sensitive Template Detection — HHpred, HMMER, and custom profile libraries identify remote homologs invisible to standard BLAST searches.
- Loop & Side-Chain Refinement — MD-based relaxation and rotamer optimization correct template-induced distortions in binding pockets.
- Ligand-Guided Model Optimization — When co-crystal structures of homologs exist, bound-ligand conformations template active-site geometry, improving docking accuracy by 30–50%.
What We Offer: For targets lacking direct homologs, our threading pipelines identify fold relationships across protein families, enabling structure-based design where sequence-based methods fail. Every model ships with per-residue confidence scores and a validation report ready for SBDD deployment.
Explore Homology Modeling →Ab Initio Modeling & Co-evolutionary Analysis
Folding Orphan Targets Without Templates
Key Features:
- Co-evolutionary Contact Prediction — GREMLIN, CCMPred, and deep learning contact maps extract structural constraints from sequence covariance, enabling accurate folding even for orphan proteins.
- Fragment Assembly & Refinement — Rosetta fragment insertion combined with MD relaxation generates physically realistic conformations.
- Pocket Prediction on De Novo Folds — FPocket and FTMap scan predicted surfaces for druggable cavities, turning sequence-only targets into virtual screening candidates.
What We Offer: For first-in-class targets with no known homologs, this is often the only path to a structural starting point. We deliver ranked structural ensembles with pocket annotations, enabling hit identification campaigns to proceed in parallel with experimental structure determination.
Explore Ab Initio Modeling →Structure Refinement & Quality Assessment
Ensuring Models Are Ready for Drug Design
Key Features:
- Comprehensive Validation Metrics — MolProbity clash scores, Ramachandran distributions, rotamer outliers, and EMRinger scores for Cryo-EM models.
- B-Factor & Local Resolution Refinement — Real-space refinement in Phenix/Coot with local resolution filtering for Cryo-EM maps; anisotropic B-factor modeling for high-resolution X-ray data.
- Stability Validation — Microsecond-scale MD tests structural integrity under thermal stress; unstable regions are flagged for re-refinement or experimental reprocessing.
What We Offer: A model with poor geometry generates false-positive docking hits and wasted synthesis cycles. Our quality gates ensure every coordinate file is drug-design-ready, with validation statistics that satisfy peer-review and regulatory scrutiny.
Explore Structure Refinement →Structural Data Processing Services
From Raw Data to Atomic Coordinates

Key Features:
- X-ray Crystallography Pipeline — HKL2000/XDS data reduction, Phaser/Molrep molecular replacement, ARP/wARP or Phenix AutoBuild model building, and REFMAC5/Phenix refinement. We handle anomalous phasing (SAD/MAD) and native data alike.
- Cryo-EM/ET Single-Particle Analysis — RELION 4.0 / cryoSPARC v4.0 motion correction, CTF estimation, particle picking (Topaz/DeepPicker), 2D classification, 3D reconstruction, and local resolution estimation. Sub-tomogram averaging for in situ structures.
- AI-Assisted Model Building — DeepTracer, Phenix map-to-model, and AlphaFold2 seeding accelerate model building in low-resolution maps, cutting manual tracing time by 60%+.
What We Offer: For pharma teams with in-house data collection, we operate as your remote structure solution center — turning diffraction images or movie stacks into deposition-ready coordinates. For biotechs, this means accessing synchrotron-grade data processing without maintaining a structural bioinformatics team.
Explore Data Processing →Binding Pocket & Druggability Analysis
Turning Structures into Actionable Targets
Key Features:
- Pocket Detection & Characterization — FPocket, Concavity, and SiteMap identify orthosteric, allosteric, and cryptic pockets; pocket volume, hydrophobicity, and hydrogen-bond potential are quantified.
- Druggability Scoring — DrugScore, FTMap hot-spot analysis, and MD-based pocket stability assessments rank pockets by ligandability.
- Cryptic Pocket Discovery — Apo/holo MD simulations reveal transient pockets invisible in static crystal structures, opening new chemical space for "undruggable" targets.
What We Offer: Every predicted or experimental structure is useless without a validated binding site. Our pocket analysis transforms raw coordinates into prioritized virtual screening targets, with druggability reports that guide library design and medicinal chemistry strategy.
Explore Druggability Analysis →Complex Structure Prediction
Modeling Multi-Component Assemblies
Key Features:
- Protein-Protein Docking — HADDOCK, ClusPro, and ZDOCK generate ranked complex poses; MD refinement validates interface stability.
- Peptide-Protein Interaction Modeling — Flexible peptide docking with induced-fit protocols; macrocycle and stapled-peptide conformational sampling.
- Nucleic Acid-Protein Complexes — Specialized force fields and MD protocols for DNA/RNA-binding proteins, including transcription factors and CRISPR complexes.
What We Offer: For PPI targets and large assemblies where experimental structures are scarce, our computational assembly pipelines provide testable models for fragment-based screening and biophysical assay design. Every complex model ships with interface scores and MM/PBSA binding free energy estimates.
Explore Complex Prediction →Antibody Structure Prediction
AI-Driven Antibody Modeling for Biologics Discovery
Key Features:
- Fv/VH-VL Orientation Prediction — IgFold and DeepAb predict antibody variable domain structures with CDR loop accuracy approaching experimental quality for canonical classes.
- CDR H3 Refinement — MD-based loop remodeling and ensemble docking correct H3 predictions for non-canonical sequences.
- Antibody-Antigen Docking — Protein-protein docking with antibody-specific scoring; epitope constraint mapping from mutagenesis or HDX-MS data.
What We Offer: For biotechs building antibody pipelines without structural biology infrastructure, we deliver humanization-ready models, affinity maturation guidance, and developability risk flags — all from sequence alone.
Explore Antibody Prediction →Technology Platform
Integrated Discovery Infrastructure: Dry Lab + Wet Lab, Zero Handoffs
Our integrated platform spans AI-driven structure prediction and experimental data processing — enabling end-to-end structural biology without the delays, IP leaks, and miscommunication that come from coordinating multiple vendors.
Computational Platform — Dry Lab
Powered by our MagHelix™ CADD Platform and MagHelix™ AIDD Platform
| Capability | Details |
|---|---|
| AI/ML Structure Engine | AlphaFold2, RoseTTAFold, IgFold, DeepAb; proprietary MSA optimization and template curation pipelines |
| Ab Initio & Threading | Rosetta, I-TASSER, HHpred; co-evolutionary contact prediction (GREMLIN/CCMPred/DeepPotential) |
| Molecular Dynamics | GROMACS/AMBER microsecond all-atom simulations; membrane protein-lipid systems; enhanced sampling (metadynamics, REST2) |
| Structure-Based Docking | Glide, AutoDock-GPU; induced-fit and covalent docking enabled |
| ADMET Prediction | Deep learning panel: Fsp3, LogP, hERG, CYP450, Papp, BBB permeability |
| Data Processing Software | Phenix, CCP4, RELION 4.0, cryoSPARC v4.0, DeepTracer, EMAN2, Coot |
Experimental Validation Platform — Wet Lab
Powered by our MagHelix™ Structural Biology and SBDD Platform
| Capability | Details |
|---|---|
| X-ray Crystallography | Co-crystal soaking, automated screening robots, high-throughput ligand soaking |
| Cryo-Electron Microscopy | Thermo Fisher Glacios / Krios G4; single-particle analysis for large complexes and membrane proteins |
| NMR Spectroscopy | Bruker 600MHz+; STD-NMR for fragment validation; protein structure determination |
| Gene-to-Protein | Codon optimization, multi-system expression, purification, and quality control |
Platform Edge: For difficult targets, the ability to pivot from AI prediction to Cryo-EM validation to co-crystal soaking without changing project teams protects your timeline and budget.

Thermo Scientific Krios G4

Rigaku XtaLAB Synergy-R

Bruker Avance NEO
Platform specifications are subject to continuous upgrade. Contact our team for instrument availability and project-specific capability assessment.
Closed-Loop Discovery Engine
When Prediction Meets Validation
Traditional structural biology separates computational prediction from experimental validation — creating handoff delays and information loss. Our platform feeds every experimental result back into the AI models.
AI Structure Prediction
AlphaFold2/RoseTTAFold generate initial models from sequence; low-confidence regions flagged for experimental focus.
→ Feeds into Wet-lab
Experimental Data Processing
Cryo-EM/X-ray structures refine predicted coordinates, updating loop conformations and side-chain rotamers.
→ Feeds into Models
Pocket & Druggability Update
Experimental structures reveal cryptic pockets and allosteric sites invisible to prediction, updating docking and screening protocols.
→ Feeds into CADD
Binding Data Feedback
SPR/ITC binding data and co-crystal structures retrain target-specific scoring functions for the next design cycle.
→ Feeds back into AI
Industrial Value:
For Biotechs
Your Phase 0 structural data becomes training data for Phase 1 targets. You don't just buy a service — you invest in a compounding learning partnership.
For Pharma
Every computational prediction is linked to an experimental outcome with project ID, timestamp, and model version — fully audit-ready for regulatory submissions and internal portfolio reviews.
Project Management & Execution
Project Workflow
A standardized, milestone-driven execution system. From sequence intake to validated structure delivery — managed by a single project team, tracked in real time.


01 Strategy
- Target sequence review, druggability assessment, and MSA curation
- Modality selection: AI prediction, homology modeling, or experimental structure determination
Deliverable: Project proposal with Gantt-chart milestones, budget, and risk matrix
02 In Silico
- AlphaFold2/RoseTTAFold prediction or homology modeling / ab initio folding
- Pocket detection and druggability scoring; docking validation
Deliverable: Predicted structural models (PDB format) with pLDDT/pTM scores and pocket annotations
03 Wet lab
- Gene-to-protein expression and purification (if experimental validation required)
- X-ray / Cryo-EM / NMR data collection and processing
Deliverable: Experimental data processing report with statistics and electron density maps
04 Validation
- Biophysical validation: SPR, TSA, DLS for protein quality
- Structure refinement and quality assessment (MolProbity, EMRinger)
Deliverable: Validated structure with refinement statistics and deposition-ready coordinates
05 Deliverables
- Complete structural report with confidence scores, validation statistics, and docking-ready coordinates
- Binding pocket analysis and virtual screening recommendations
Deliverable: Final technical report + electronic data package + Hit-to-Lead / Hit ID transition plan
Sample Requirements
| Sample Type | Specification |
|---|---|
| Target Sequence | FASTA format; species source; domain boundaries (if known); post-translational modification sites |
| Reference Structures | PDB IDs or experimental data of homologs (if available; not required for ab initio) |
| Experimental Data (for processing services) | Diffraction images (X-ray), movie stacks (Cryo-EM), or spectral data (NMR) with collection parameters |
Standard Deliverables
Upon project completion, clients receive comprehensive reports including:
- Predicted or refined structural coordinates (PDB format) with confidence metrics
- Validation statistics (MolProbity scores, Ramachandran plots, EMRinger scores)
- Binding pocket analysis with druggability scores and prioritized sites
- Computational docking models and virtual screening recommendations
- ADMET risk flags for pocket-compatible chemotypes
- Follow-up optimization recommendations and project retrospectives
Our technical team responds within 24 hours. All inquiries protected under NDA.
Frequently Asked Questions
Case Study
Case #1
X-Ray Crystallography of XYZ Protein–DNA/RNA Complex
Goal: Resolve the 3D crystal structure of a protein–nucleic acid complex to enable structure-guided design for non-traditional target classes.
Key Data:
- Resolution: 2 Å; Space group I222 (a = 80.330 Å, b = 82.423 Å, c = 126.061 Å)
- Optimized condition: 0.15 M CaCl₂, 0.1 M NaAc, pH 5.0, 40% MPD (8-week incubation, reproducible)
- Phasing strategy: Molecular replacement via HKL3000 using PDB 4MH8 dual-domain split (T24–I261 + Q263–T482) with nucleic acid component
Why it matters: Nucleic acid–protein complexes are high-value, high-difficulty targets that standard CRO workflows avoid. This validates our end-to-end structural data processing pipeline — from recombinant expression and complex reconstitution to high-throughput screening, synchrotron data collection, and advanced phasing for problematic MR scenarios. For biotechs, it means accessing beamline-grade X-ray crystallography data analysis without the CapEx. For pharma, it means a single accountability chain delivering deposition-ready coordinates from non-traditional targets.

Figure 1. XYZ-DNA/RNA complex crystals in different crystallization solutions.

Figure 2. XYZ-DNA/RNA complex crystal diffraction data.
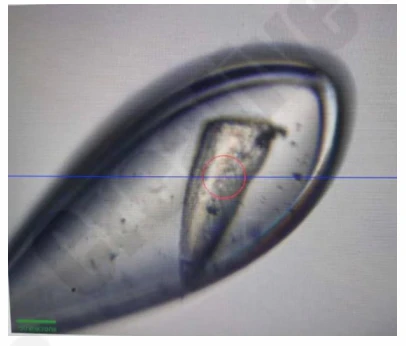
Figure 3. XYZ-DNA/RNA complex crystals in optimized crystallization solutions.
Case #2
Particle Analysis of Recombinant AAV by Cryo-EM
Goal: Quantify empty/full particle ratios and validate capsid morphology of a recombinant adeno-associated virus (AAV) sample to support gene therapy CMC and IND-enabling documentation.
Key Data:
- Particle statistics: 2,703 particles analyzed across 44 Cryo-TEM images; 77.6% empty, 17.8% full, 4.6% uncertain — classified by radial electron density and circularity via Mempro auto-detection with manual curation
- Instrumentation: FEI Talos F200C (200 kV) Cryo-TEM; sample quality pre-assessed by negative-stain TEM (FEI Tecnai Spirit, 120 kV) confirming no aggregation and optimal concentration without dilution
- Structural fidelity: All particles exhibit icosahedral symmetry; dodecahedral nucleocapsid structure clearly resolved at 45k magnification; full particles show distinct internal electron density with no shell-core boundary overlap
Why it matters: For gene therapy developers, empty AAV capsids are process-related impurities that reduce potency and trigger regulatory scrutiny. This case demonstrates our Cryo-EM single-particle analysis capability applied to biopharmaceutical QC — delivering quantitative empty/full ratios and morphology verification with full particle-level traceability. For biotechs advancing AAV vectors toward IND, this means CMC-grade data without electron microscopy infrastructure. For pharma, this means batch-release analytics and regulatory documentation from a single analytical partner.

Figure 1. Examination of sample quality by negative staining TEM.

Figure 2. Characterization of AAV particles by cryo-TEM.
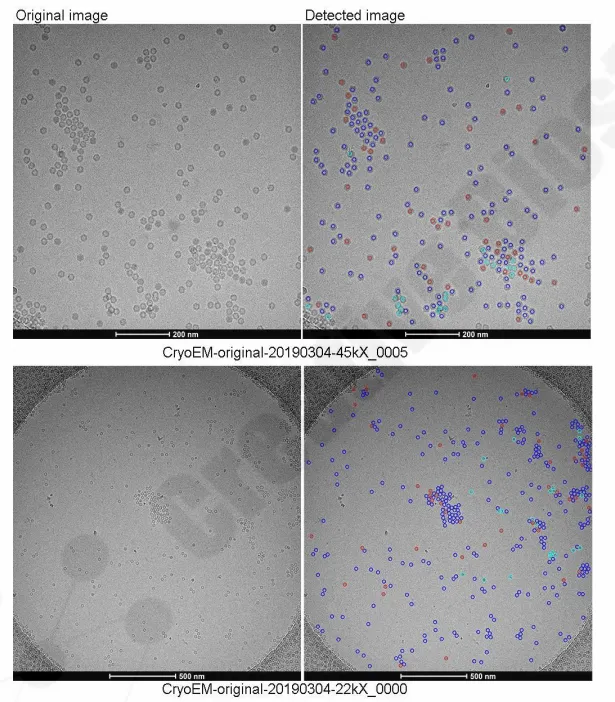
Figure 3. Particle classes recognized by MemproTM image processing software.