Molecular Docking Services
Docking without physical validation generates false positives. We deliver AI-generated poses refined by force-field minimization and MD — scoring with CNN ensembles and validating through co-crystallization to ensure chemically plausible binding modes.
Why Molecular Docking Is the Critical Foundation
Structure-based hit identification without reliable docking is screening without a filter. Seed-stage biotechs need to triage millions of compounds overnight; pharma teams require pose accuracy that survives FEP and medicinal chemistry scrutiny. Our platform combines diffusion-model pose generation, force-field refinement, and CNN scoring — then validates top hits through X-ray or Cryo-EM to eliminate the false positives that plague pure-AI workflows.
What Sets the Platform Apart
AI + Physics Hybrid
Diffusion models generate poses in seconds; force-field minimization resolves steric clashes and optimizes H-bonds. Physics ensures chemical realism.
Cross-Docking Validated
Rigid, flexible, and induced-fit protocols are benchmarked against cross-docking datasets — not just self-docking — ensuring performance on novel chemistries.
Covalent-Ready
Covalent docking protocols map reactive cysteines, serines, and lysines; warhead compatibility is scored before synthesis.
Technology Suite
Protein-Ligand Docking (Rigid / Flexible / Induced Fit Docking)
High-throughput pose generation with multi-level receptor flexibility
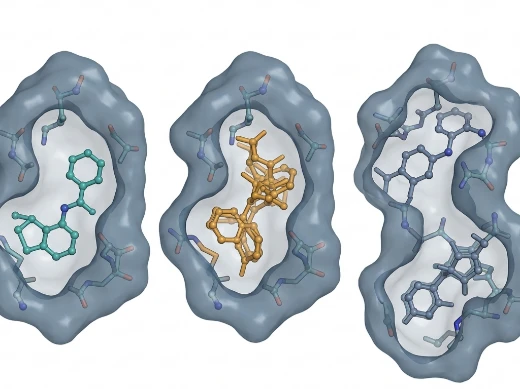
Key Features:
- Rigid Docking — High-throughput pose generation for large library screening with fixed receptor conformations; optimized for initial hit identification.
- Flexible Docking — Side-chain rotamer sampling and ligand conformational search for refined binding mode prediction.
- Induced Fit Docking — Full receptor backbone and side-chain flexibility to capture binding-pocket remodeling upon ligand engagement.
Ideal For: Seed-stage biotechs requiring rapid hit triage; pharma teams seeking accurate pose prediction for lead optimization; programs targeting flexible binding pockets (kinases, nuclear receptors).
What We Offer:
Parallel-path docking: rigid screening for speed, flexible and induced-fit protocols for precision. Deliverables include ranked pose libraries, interaction maps, and MD-refined binding modes.
Protein-Protein/Peptide Docking (Rigid Body / Flexible Docking)
PPI complex assembly and interface refinement
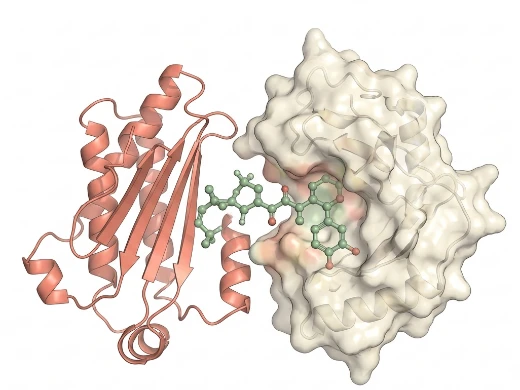
Key Features:
- Rigid Body Docking — Fast geometric matching for PPI complex assembly using ZDOCK/FtMap-style algorithms; ideal for initial landscape mapping.
- Flexible Docking — HADDOCK/RosettaDock protocols integrating experimental restraints (crosslinking, mutagenesis) to refine interface geometry.
- Peptide-Specific Sampling — Specialized force fields and peptide conformational libraries for short linear motif recognition.
Ideal For: Transient PPI target programs; peptide therapeutic discovery; antibody-antigen complex modeling.
What We Offer:
From coarse-grained PPI mapping to atomistic interface refinement. Restraint-driven protocols leverage your existing SAR or biophysical data to constrain the search space and improve accuracy.
Protein-Nucleic Acid Interaction Modeling (Docking)
DNA/RNA groove recognition and non-canonical structure targeting
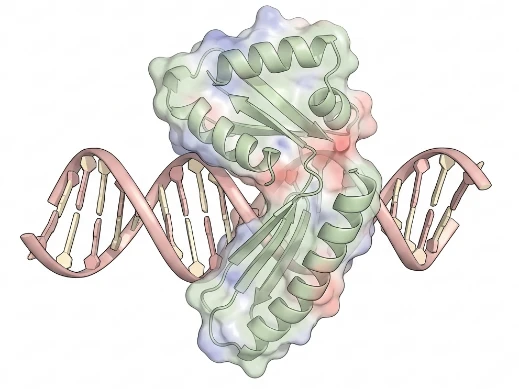
Key Features:
- DNA/RNA Groove Recognition — Docking protocols optimized for major/minor groove binding and intercalation modes.
- G-Quadruplex & Riboswitch Targeting — Specialized scoring functions for non-canonical nucleic acid structures.
- Protein-DNA Crosslinking Integration — Incorporation of XL-MS and FRET restraints to guide nucleoprotein complex assembly.
Ideal For: Transcription factor inhibitor programs; RNA-targeting small molecules; CRISPR effector complex engineering.
What We Offer:
Nucleic acid docking remains underrepresented in commercial platforms. We deliver validated protocols for DNA/RNA-binding proteins, with MD refinement to capture electrostatic and desolvation effects absent in static scoring.
Covalent Docking
Warhead-specific libraries and reaction-coordinate sampling
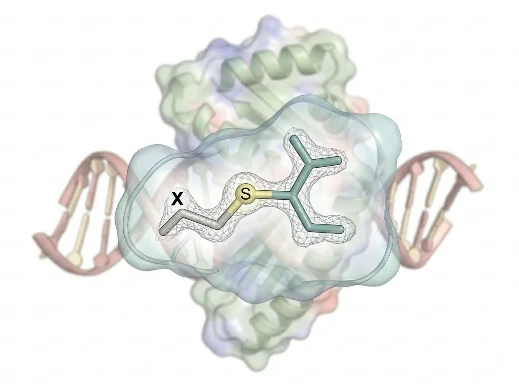
Key Features:
- Warhead-Specific Libraries — Pre-parameterized reactive groups (acrylamides, vinyl sulfones, boronic acids, nitriles) with bond-forming energy terms.
- Reaction-Coordinate Sampling — Conformational search that includes covalent bond formation geometry and transition-state mimicry.
- Selectivity Profiling — Proteome-wide covalent liability scanning against cysteine, lysine, and serine nucleophiles.
Ideal For: Cysteine-targeted covalent inhibitors; PROTAC warhead optimization; irreversible kinase inhibitor programs.
What We Offer:
Covalent docking requires specialized force fields and careful warhead parameterization. We deliver pose predictions with covalent bond geometry validation, followed by MD simulation to confirm conjugate stability and selectivity.
Scoring Functions & Post-docking Refinement
Physics-based and ML-enhanced rescoring for accurate affinity ranking
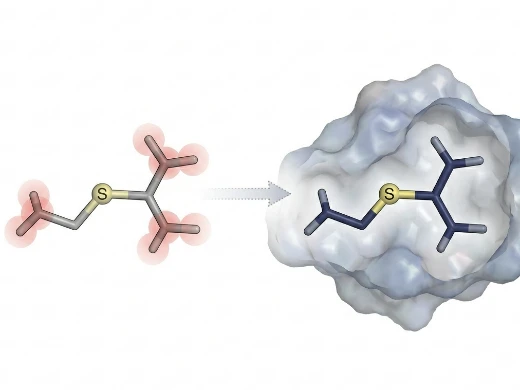
Key Features:
- Physics-Based Scoring — MM/PBSA, MM/GBSA, and FEP rescoring for accurate affinity ranking beyond docking score approximations.
- ML-Enhanced Scoring — RTMScore, EquiScore, and proprietary classifiers trained on 500+ co-crystal structures to improve enrichment in virtual screening.
- Post-Docking Optimization — Force-field relaxation (FF) and alignment-based refinement (Align) to resolve steric clashes and chirality errors from AI docking poses.
Ideal For: Large-scale VS campaigns requiring enrichment optimization; programs where docking scores alone fail to correlate with measured affinity.
What We Offer:
A hierarchical scoring strategy: fast docking scores for initial triage, physics-based and ML rescoring for final ranking, and post-docking refinement to ensure physical plausibility of AI-generated poses.
Platform Instrumentation
Core Instruments & Software
| Instrument / Software | Capability |
|---|---|
| NVIDIA DGX A100 | Diffusion-model inference and large-scale virtual screening |
| NVIDIA RTX A6000 Cluster | Real-time pose visualization and ensemble analysis |
| GNINA 1.3 / AutoDock Vina | CNN-scored and physics-based docking at scale |
| Schrödinger Glide / IFD-MD | Induced-fit docking and Prime refinement |
| OpenEye / RDKit | Ligand preparation, tautomer enumeration, and pose analysis |
| GROMACS/AMBER HPC | Post-docking MD relaxation and FEP validation |
Standardized Workflow
Project Workflow
A milestone-driven execution system from target to prioritized hits.


01 Target Review
- Pocket analysis and druggability assessment
- Ligand library curation
- Deliverable: Target assessment report + docking protocol
02 AI Docking
- AI pose generation (DiffDock/GNINA)
- Ensemble docking across MD conformers
- Deliverable: Raw pose ensemble + confidence scores
03 Physics Refinement
- Force-field minimization
- Steric clash and chirality correction
- Deliverable: Refined poses + quality metrics
04 Hit Score
- CNN ensemble scoring
- FEP/MM-PBSA ranking (optional)
- Deliverable: Prioritized hit list + binding mode report
05 Validation
- Co-crystal soaking or Cryo-EM validation (optional)
- Deliverable: Validated structures + final report
Sample Requirements
- Target structure: PDB file, AlphaFold model, or homology model
- Ligand library: SMILES, SDF, or curated compound set (up to millions)
- Known binders: Reference compounds for pose validation and benchmarking
- Project scope: Target class, desired selectivity profile, and covalent vs. non-covalent requirement
- Restraints: SAR data, mutagenesis, or crosslinking information (if available)
Standard Deliverables
- Docked pose ensemble with AI confidence and physics-based scores (PDB/SDF)
- Binding mode analysis with interaction fingerprints and hydrogen-bond maps
- Refined poses post-minimization with quality checks (PoseBusters/PoseCheck)
- FEP or MM-PBSA ranking report (if selected)
- Experimental validation data (if selected): X-ray or Cryo-EM coordinates
- Final technical report with hit prioritization and SAR recommendations
Frequently Asked Questions
Case Study
Case Study: PoseX — AI Defeats Physics on Protein-Ligand Cross Docking, But Hybrid Wins
Goal: Benchmark 23 docking methods across self-docking and cross-docking scenarios to establish the optimal AI/physics strategy for industrial virtual screening.
Key Data:
- Benchmark scale: 718 self-docking + 1,312 cross-docking entries spanning diverse protein families and ligand chemistries.
- AI superiority: AI docking methods (DiffDock, ArtiDock, Uni-Mol) consistently outperform physics-based methods (Glide, Vina) in overall success rate.
- Hybrid dominance: AI-generated poses followed by physics-based relaxation achieve the highest accuracy — combining AI search efficiency with force-field validity.
- Chirality fix: AI co-folding methods exhibit stereochemistry hallucinations; physics-inspired potentials (Boltz-1x) resolve these, confirming the necessity of hybrid workflows.
Why it matters: For drug developers running large-scale virtual screens, this study validates that pure-AI docking risks physical implausibility, while pure-physics methods miss optimal poses. By pairing diffusion-model generation with force-field refinement, our platform delivers both speed and chemical accuracy — directly supporting hit identification for targets where traditional docking fails.
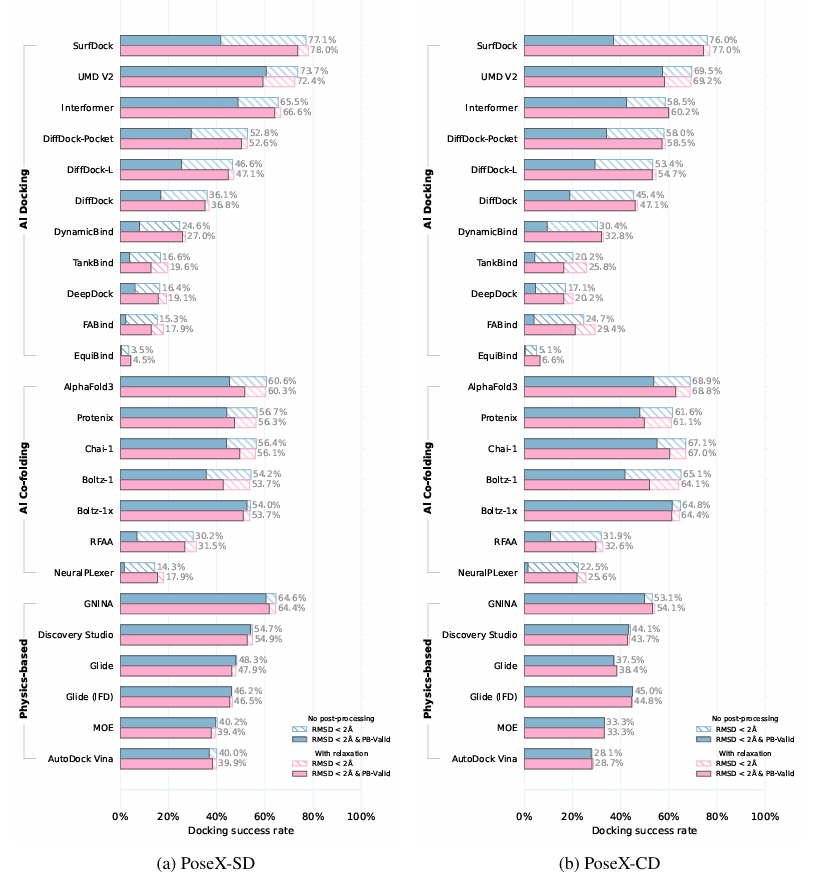
Figure 1. Performance on PoseX-SD and PoseX-CD datasets showing proportions of predictions with RMSD < 2 Å (striped bars) and PoseBuster-validated results (solid bars), comparing outcomes with and without structural relaxation. (Jiang Y.; et al. 2025)
Reference
Jiang Y, et al. PoseX: AI Defeats Physics Approaches on Protein-Ligand Cross Docking. arXiv. 2025 May 3;2505.01700.
Our technical team responds within 24 hours. All inquiries protected under NDA.