AI-Enhanced Structural Analysis: Bridging the Gap Between Prediction and Atomic Truth
We redefine the MagHelix™ Gene-to-Protein and Gene-to-Structure pipeline by integrating Generative AI and Deep Learning into our world-class biophysical platforms. While AI predicts, our structural analysis provides the definitive experimental proof—delivering atomic-resolution data to validate Mechanism of Action (MOA) and secure your Intellectual Property.
Overcoming Structural Bottlenecks in Modern Drug Discovery
The transition from a computational hit to a validated lead is often stalled by the "structural bottleneck"—long crystallization timelines, the inherent flexibility of GPCRs and membrane proteins, and the high cost of experimental trial-and-error.
The "Dry-to-Wet" Synergy
Our strategic response is the AI-Enhanced MagHelix™ Platform. We use AI to "narrow the search space" through Virtual Co-crystal Screening and Automated Particle Picking, then employ our gold-standard X-ray Crystallography, Cryo-EM, and NMR facilities to provide the "Atomic Truth." This synergy ensures 100% reliable structural data for IND-enabling packages and high-stakes Structure-Based Drug Design (SBDD).
Success Rate
Reduction
Simulations Monthly
Data Turnaround
Service Portfolio: From Genetic Sequence to Atomic Precision
Our structural biology suite bridges the gap between AI-driven hypotheses and experimental truth. We provide high-fidelity validation to transform computational models into definitive structural data for regulatory success.
AI Focus: Deep learning-based codon optimization, solubility prediction, and AlphaFold-assisted domain mapping.
Our platform integrates a proprietary library of expression vectors and specialized cell lines. And we leverage AI-optimized expression strategies and MagHelix™ systems to produce high-yield, "crystal-grade" proteins (>95% purity). Our platform specializes in overcoming production bottlenecks for challenging targets like GPCRs and ion channels, ensuring optimal samples for downstream structural determination.
AI Focus: Virtual co-crystal screening, computational binding mode assessment, and AI-enhanced density refinement.
Prior to wet-lab trials, we utilize AI to evaluate ligand-binding feasibility and co-crystallization potential. Combined with synchrotron beamlines and high-throughput robotics, we rapidly transform AI-generated models into atomic-resolution experimental "truth" to support SBDD.
AI Focus: Deep learning-based particle picking (Topaz), AI-driven 3D reconstruction, and Micro-ED automation.
Our platform integrates AI to accelerate the transition from raw micrographs to high-resolution 3D density maps. By automating particle picking and real-time processing, we deliver rapid structural insights for complex membrane proteins and provide Micro-ED for small-molecule absolute configuration.
AI Focus: ML-assisted spectral assignment, chemical shift prediction, and automated fragment screening analysis.
NMR serves as the ultimate tool to validate AI-predicted protein dynamics and binding pockets in solution. We employ STD-NMR to physically verify weak-binding hits and conformational flexibility, providing a closed-loop validation for allosteric effects and protein-ligand interactions.
Structural Technology Selection Guide
This version highlights the direct economic benefits (Efficiency/Time) which are critical for decision-making.
| Technology | Ideal Target Type | AI-Enhanced Advantage | Key Deliverable | Time Savings (B2B Impact) |
|---|---|---|---|---|
| X-ray Crystallography | Small-to-medium proteins, Protein-Ligand complexes | Virtual Co-crystal Screening to prioritize high-probability soaking conditions. | Atomic-resolution (< 2.0 Å) 3D coordinates. | 40% Reduction in crystallization timelines. |
| Cryo-EM (SPA) | Large complexes (> 100 kDa), Membrane proteins (GPCRs) | Deep Learning Particle Picking to resolve structures from low-concentration samples. | High-resolution 3D density maps in native-like states. | 60% Faster data processing & 3D reconstruction. |
| NMR Spectroscopy | Small proteins (< 30 kDa), Flexible loops, IDPs | ML-assisted Spectral Assignment to rapidly map binding pockets and dynamics. | Solution-state dynamics and pKa values. | 50% Decrease in manual assignment labor. |
| Micro-ED | Small molecules, Peptides, Sub-micron crystals | Automated Diffraction Indexing for rapid absolute configuration determination. | Definitive 3D structure of micro-sized chemical leads. | 24-Hour data turnaround for lead validation. |
Workflow: From Digital Models to Atomic Truth
Our integrated pipeline synchronizes AI-driven optimization with high-resolution biophysical validation to ensure rapid and reliable structural determination.
AI-Driven Feasibility & Risk Assessment
Before initiating wet-lab trials, we utilize deep learning models to evaluate protein foldability, ligand-binding feasibility, and co-crystallization potential, effectively "narrowing the search space" and de-risking the project.
AI-Driven Construct Design & Expression Optimization
We utilize AlphaFold-assisted domain mapping and machine-learning codon optimization to design high-yield expression constructs for challenging targets, including GPCRs and membrane proteins.
Precision Protein Production & Quality Control
High-purity, "crystal-grade" proteins are produced via automated AKTA FPLC systems, with monodispersity and stability verified by DLS and MALS to ensure suitability for structural studies.
Virtual Screening & Experimental Setup
Computational binding mode assessments and virtual co-crystal screenings are performed to prioritize the most promising ligand-target combinations before initiating high-throughput X-ray or Cryo-EM trials.
High-Resolution Data Acquisition
Atomic-level data is captured through synchrotron X-ray diffraction, AI-accelerated Cryo-EM particle picking, or high-field NMR spectroscopy to visualize precise molecular interactions.
Structural Refinement & MOA Validation
Raw data is processed using AI-enhanced refinement algorithms to generate final 3D PDB models, providing definitive experimental validation for drug binding modes and mechanism of action (MOA).
Sample Requirements & Deliverables
To ensure the highest atomic resolution and data reliability, we adhere to strict quality control standards for both incoming samples and outgoing project deliverables.
Sample Requirements
We accept samples at various stages, from genetic sequences to pre-purified proteins. Our team provides buffer optimization and stability testing upon receipt.
- •
Gene/Constructs: Optimized DNA sequences in standard cloning vectors (minimum 5 μg).
- •
Purified Proteins: Minimum concentration of 1-5 mg/mL (target-dependent), with >95% purity verified by SDS-PAGE. Samples should be shipped on dry ice in optimized storage buffers.
- •
Small Molecules/Ligands: Lyophilized powder or DMSO stock (minimum 10-20 mM) with HPLC-verified purity.
- •
Complexes: For Cryo-EM or X-ray soaking, pre-formed complexes or individual components with validated binding affinity (e.g., SPR/ITC data) are preferred.
Deliverables & Data Package
Every structural analysis project concludes with a comprehensive, IND-enabling data package designed for direct integration into regulatory filings and downstream SBDD workflows.
- •
Validated 3D Structural Models: Final refined PDB files with atomic coordinates and anisotropic displacement parameters.
- •
High-Resolution Density Maps: For Cryo-EM projects, we provide unmasked and sharpened 3D volumes (mrc/map files) along with Euler angle distribution plots.
- •
Raw Experimental Data: Full sets of X-ray diffraction frames, Cryo-EM micrographs (movies), or processed NMR spectra.
- •
Comprehensive Technical Report: A detailed dossier including protein purification profiles (SEC/MALS), crystallization/vitrification conditions, data collection statistics (Table 1), and structural refinement metrics (R-factors, Ramachandran plots).
- •
Interaction Analysis: Detailed visualization of the binding pocket, hydrogen-bonding networks, and hydrophobic interactions for protein-ligand complexes.
Case Study
Case: Identification of a Functional Hydrophobic Pocket in D2 Dopamine Receptor via Computational-Experimental Synergy
Background: Signaling bias—the ability of an agonist to preferentially activate one intracellular pathway over another—is a frontier in developing GPCR-targeted drugs with fewer side effects. This study investigates the structural basis of G protein-biased signaling at the D2 dopamine receptor (D2R), demonstrating how localized structural changes in the binding pocket can allosterically drive functional selectivity.
Key Technical Milestones:
- In Silico Binding Site Prediction: Using molecular dynamics (MD) simulations and computational docking, researchers identified a unique interaction between G protein-biased ligands and a specific hydrophobic pocket. This pocket is located at the interface of the second extracellular loop (EL2) and the fifth transmembrane segment (TM5).
- Mechanistic Modeling of Signaling Bias: The study utilized computational modeling to predict that ligand interactions with this hydrophobic pocket prevent the structural rearrangements necessary for β-arrestin recruitment, while maintaining the receptor's ability to activate G proteins.
- Structure-Guided Mutagenesis Validation: To verify the "Digital Hypothesis," a series of site-directed mutations were designed based on the computational model. By altering the residues within the predicted hydrophobic pocket (e.g., I184 and V190), the team experimentally confirmed the loss of functional selectivity, proving the pocket's critical role.
- Precision Allosteric Mapping: The integration of computational analysis with pharmacological assays allowed for the precise mapping of allosteric networks within the receptor. This provides a blueprint for MagHelix™ to design "biased ligands" that target specific signaling outcomes without triggering off-target effects.
Conclusion: This research represents a significant advancement in understanding GPCR functional selectivity through the lens of structural biology. By leveraging computational modeling to identify allosteric triggers and closing the loop with experimental validation, researchers can get a sophisticated R&D framework. This synergy drastically reduces the trial-and-error in early-stage discovery, enabling the design of next-generation therapeutics with high atomic precision and tailored functional profiles.
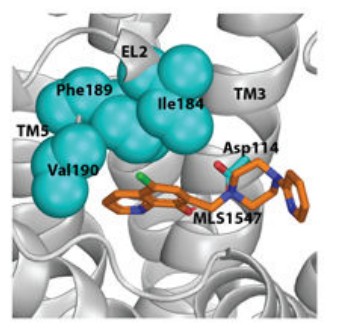
Figure 1. Atomic-level modeling of the D2R allosteric hydrophobic pocket. (Sanchez-Soto M, et al., 2020)
Why Choose Us - Our Core Advantages
Multidisciplinary Ph.D. Team
Our core scientific team consists of Ph.D. experts specialized in computational biology, medicinal chemistry, and structural biology, bringing an average of 10+ years of industrial R&D experience to every project. This collective expertise ensures that every AI-generated model is refined by deep biological insight.
State-of-the-Art AI Architecture
We integrate the latest industry-leading algorithms, including AlphaFold-3 and RFdiffusion, with our proprietary MagHelix™ platforms. This allows us to provide unparalleled predictive accuracy for complex systems, including multi-protein complexes, nucleic acid interactions, and post-translational modifications.
High-Performance Computing Infrastructure
Our specialized GPU clusters and optimized parallel processing workflows ensure rapid turnaround times for large-scale virtual screening and microsecond-scale MD simulations. This infrastructure allows us to handle high-throughput structural analysis without compromising on data resolution or accuracy.
Integrated "Dry-to-Wet" Validation
Unlike pure computational firms, we provide a seamless transition to wet-lab validation. Utilizing X-ray crystallography, Cryo-EM, and SPR, we experimentally confirm in silico leads and provide the "atomic truth" required to de-risk your clinical pipeline and support regulatory filings.

Frequently Asked Questions
For any inquiries, our drug discovery experts are ready to help you get technical support to accelerate your hit-to-lead transition and maximize the success rate of your preclinical candidates.