Molecular Dynamics (MD) Simulations
Molecular dynamics reveals what crystallography cannot — cryptic pockets, induced-fit binding, and allosteric signaling. We run microsecond simulations, FEP free energy, and membrane systems, all experimentally calibrated under one roof.
Creative Biostructure at a Glance
Over a decade of trusted expertise powering biotech, pharma, and research institutions worldwide to advance therapeutic innovation.
Why Partner With Us
Static docking scores assume rigid receptors. In reality, proteins breathe, pockets collapse, and allosteric networks rewire on the nanosecond scale. Teams that ignore dynamics burn synthesis cycles on compounds that look perfect in a crystal structure but fail in solution because the binding mode was never thermodynamically stable. We built this platform to replace snapshot-based intuition with time-resolved, physics-based certainty — every simulation is designed to answer a specific medicinal chemistry question, and every prediction is calibrated against experimental binding data.
Your CapEx is in chemistry and disease biology. Ours is in HPC infrastructure and force-field expertise.
| Stage | What We Deliver | What You Don't Need to Build |
|---|---|---|
| System Preparation | AI-predicted or experimental structure equilibration; ligand parameterization; membrane bilayer assembly; solvent and ion neutralization | HPC cluster, force-field curation team |
| Simulation Execution | Microsecond all-atom MD; enhanced sampling (aMD, metadynamics, REST2); coarse-grained modeling for large assemblies | GPU farm, GROMACS/AMBER/NAMD licenses |
| Free Energy Prediction | FEP/TI relative binding affinity; MM/PBSA end-point scoring; selectivity profiling across homolog panels | Alchemical calculation infrastructure |
| Experimental Calibration | SPR/ITC validation of predicted ΔG; Cryo-EM conformational state comparison; TSA thermal stability verification | Biophysics suite, structural biology team |
Production-Ready Deliverables: Every simulation ships with trajectory analysis reports, FEP-derived ΔΔG values, conformational ensemble clustering, and binding free energy decomposition — your medicinal chemists can prioritize synthesis with thermodynamic rationale, or our Lead Optimization team can advance the program.
- ✓ Milestone-based pricing aligned with your fundraising cycles
- ✓ No HPC maintenance overhead — simulation design, execution, and analysis under a single project manager
Membrane proteins. Intrinsically disordered regions. Allosteric modulation. Conformational selection. These are phenomena that only dynamics can explain.
Proven track record on flexible targets
GPCRs, ion channels, and nucleic acid-protein complexes where static structures mislead. Our membrane protein-lipid MD captures lipid-driven gating and allosteric coupling invisible to crystallography.
Enhanced sampling for rare events
Accelerated MD, REMD, and metadynamics access millisecond-scale conformational transitions on microsecond budgets, revealing cryptic pockets and allosteric pathways for fragment-based and covalent design.
Thermodynamic precision for lead optimization
FEP calculations predict relative affinity changes before synthesis, directing R-group modifications toward enthalpically or entropically favorable vectors — validated by ITC and SPR without vendor handoffs.
Core Service Modules
Service Module At-a-Glance
| Service | Core Capability | Structural + Computational Integration | Typical Timeline |
|---|---|---|---|
| Binding Free Energy Calculation | FEP/TI relative affinity; MM/PBSA end-point scoring; selectivity profiling | Co-crystal structures or Cryo-EM maps seed simulation starting conformations; SPR/ITC calibrates FEP parameters | 3–6 weeks |
| All-Atom Protein MD Simulation | Microsecond explicit-solvent trajectories; conformational sampling; stability and flexibility profiling | AlphaFold2 or homology models provide starting structures; docking poses are equilibrated and scored dynamically | 2–4 weeks |
| Membrane Protein & Lipid MD Simulation | Explicit lipid bilayer assembly; gating dynamics; lipid-protein interaction mapping; membrane insertion analysis | Cryo-EM density maps guide bilayer orientation; TSA validates thermal stability predictions | 4–8 weeks |
| Nucleic Acid MD Simulation | DNA/RNA flexibility and bending; protein-nucleic acid interaction dynamics; groove binding and intercalation analysis | Complex structure prediction provides initial poses; NMR NOE restraints refine trajectories | 3–6 weeks |
| Replica Exchange MD (REMD) | Parallel temperature replicas for enhanced conformational sampling; free energy landscape mapping; rare event access | Co-crystal or Cryo-EM states seed replica starting points; docking and pharmacophore models are validated against sampled ensembles | 4–8 weeks |
| Coarse-Grained (CG) MD Simulation | Martini force-field large assemblies; membrane remodeling; protein-protein association kinetics; millisecond-scale events | All-atom MD benchmarks CG parameters; Cryo-ET validates large-scale morphological predictions | 3–5 weeks |
Binding Free Energy Calculation
FEP-Guided Synthesis Prioritization

Key Features:
- Alchemical Free Energy Perturbation (FEP/TI) — GROMACS/AMBER GPU-accelerated relative binding affinity calculations predicting ΔΔG within ~1 kcal/mol of experimental SPR/ITC data for well-calibrated targets.
- MM/PBSA End-Point Scoring — Post-trajectory free energy decomposition into enthalpic and entropic contributions, identifying whether affinity gains are driven by H-bond networks or hydrophobic burial.
- Selectivity Profiling — FEP calculations across homolog panels (kinase family, GPCR subtypes) flag off-target risks before synthesis commitment.
What We Offer: For lead optimization programs, FEP replaces intuition with thermodynamic certainty — you synthesize only modifications with predicted affinity improvements. For biotechs without alchemical calculation infrastructure, we deliver ΔΔG rankings with biophysical validation to confirm directionality.
Explore FEP Calculations →All-Atom Protein MD Simulation
Microsecond Trajectories for Drug-Design-Ready Insights
Key Features:
- Explicit-Solvent Microsecond Sampling — GROMACS/AMBER/NAMD with AMBER ff19SB or CHARMM36m force fields; particle-mesh Ewald electrostatics; reproducible trajectory protocols.
- Induced-Fit and Cryptic Pocket Discovery — Trajectory analysis reveals transient pocket openings and loop rearrangements invisible in crystal structures, updating docking and virtual screening protocols.
- Ligand-Binding Stability Assessment — Docking poses are equilibrated in explicit solvent to test whether predicted binding modes survive thermal fluctuations and solvent competition.
What We Offer: For seed-stage biotechs, dynamic validation of docking poses before spending on synthesis. For pharma, conformational ensemble data that supports regulatory filings and intellectual property claims around novel binding modes.
Explore All-Atom MD →Membrane Protein & Lipid MD Simulation
Capturing Lipid-Driven Biology
Key Features:
- Explicit Lipid Bilayer Assembly — Automated bilayer construction (POPC, POPE, cholesterol mixtures) with protein orientation guided by Cryo-EM density or OPM database; hydration and ionic strength matched to experimental conditions.
- Gating and Allosteric Coupling — Microsecond trajectories reveal lipid-mediated conformational transitions, sodium/proton coupling pathways, and allosteric signal propagation in GPCRs, ion channels, and transporters.
- Ligand Entry and Membrane Partitioning — Enhanced sampling (steered MD, metadynamics) maps ligand entry pathways from bulk solvent through the bilayer to the orthosteric site.
What We Offer: For membrane protein programs where crystallization fails and Cryo-EM provides only a few states, MD reveals the dynamic landscape connecting those states — critical for understanding activation mechanisms and designing state-selective modulators.
Explore Membrane MD →Nucleic Acid (DNA/RNA) MD Simulation
Dynamic Models for Non-Traditional Targets
Key Features:
- DNA/RNA Flexibility and Bending — Explicit-solvent trajectories capturing major/minor groove width fluctuations, base-pair opening events, and backbone conformational transitions.
- Protein-Nucleic Acid Interaction Dynamics — Simulation of transcription factor binding, CRISPR R-loop formation, and helicase translocation along nucleic acid substrates.
- Groove Binding and Intercalation Analysis — Trajectory-derived binding free energy profiles for small molecules targeting DNA/RNA grooves, including sequence-specific selectivity.
What We Offer: For nucleic acid-targeting programs where static structures provide incomplete pictures of ligand accessibility, MD reveals transient groove conformations and competitive ion effects that determine binding selectivity.
Explore Nucleic Acid MD →Replica Exchange Molecular Dynamics (REMD)
Accessing Millisecond Events on Microsecond Budgets
Key Features:
- Parallel Temperature Replicas — Multiple simultaneous simulations at different temperatures exchange conformations, accelerating escape from kinetic traps and sampling rare states.
- Free Energy Landscape Mapping — Cluster analysis and principal component analysis of REMD trajectories reveal low-energy conformational basins and transition pathways.
- Rare Event Access — Cryptic pocket opening, loop folding, and allosteric switching events that standard MD cannot reach within accessible timescales.
What We Offer: For targets with known inactive and active states but unclear transition mechanisms, REMD provides thermodynamic and kinetic landscapes that guide the design of state-selective allosteric modulators and covalent traps.
Explore REMD →Coarse-Grained (CG) MD Simulation
Millisecond-Scale Assembly and Membrane Remodeling
Key Features:
- Martini Force-Field Large Assemblies — Protein complexes, viral envelopes, and lipid nanoparticles simulated at ~1000× speedup over all-atom, accessing biologically relevant timescales.
- Membrane Remodeling and Fusion — Bilayer curvature, pore formation, and vesicle fusion events relevant to drug delivery and membrane protein biogenesis.
- Protein-Protein Association Kinetics — CG simulations predict encounter rates and complex lifetimes for PPI targets too large for all-atom approaches.
What We Offer: For large-scale systems where all-atom MD is computationally prohibitive, CG provides morphological and kinetic insights at experimental timescales, validated against Cryo-ET and light-sheet microscopy.
Explore CG MD →Technology Platform
Integrated Dynamics Infrastructure: HPC Simulation + Experimental Calibration, Zero Handoffs
Computational Platform — Dry Lab
Powered by our MagHelix™ CADD Platform and dedicated HPC cluster
| Capability | Details |
|---|---|
| GPU-Accelerated MD Engine | GROMACS 2023+ / AMBER 22+ / NAMD 3.0+; CUDA-enabled for NVIDIA A100/H100; microsecond/day throughput for ~100k-atom systems |
| Alchemical Free Energy | GROMACS/AMBER GPU FEP and Thermodynamic Integration (TI); relative and absolute binding free energy protocols |
| Enhanced Sampling | Metadynamics, REST2, umbrella sampling, steered MD, accelerated MD (aMD); custom collective variable design for cryptic pocket and allosteric pathway access |
| Membrane Systems | CHARMM-GUI, PACKMOL, and in-house bilayer assembly pipelines; lipid parameter libraries (POPC, POPE, cholesterol, custom mixtures) |
| Force-Field Curation | AMBER ff19SB, CHARMM36m, OPLS-AA; ligand parameterization via GAFF2, CGenFF, and QM-derived torsional profiles |
| Trajectory Analysis | MDTraj, MDAnalysis, PyEMMA; Markov state modeling; principal component and cluster analysis |
Platform Edge: The ability to run FEP on Monday, synthesize the top-ranked derivative on Wednesday, and validate with ITC by Friday — all under one project team — compresses traditional 3-month optimization loops into 2-week iterations.
Experimental Calibration Platform — Wet Lab
Powered by our MagHelix™ Structural Biology and SBDD Platform
| Capability | Details |
|---|---|
| SPR/ITC | Biacore 8K+ / MicroCal PEAQ-ITC for experimental ΔG calibration and FEP validation |
| Cryo-EM | Thermo Fisher Krios G4 for conformational state validation against simulated ensembles |
| NMR | Bruker 600MHz+ for dynamic order parameters and NOE-restrained ensemble validation |
| Thermal Stability | Prometheus NT.48 DSF / DSC for simulation-predicted melting and aggregation validation |
Wet Lab Edge: The ability to run FEP on Monday, synthesize the top-ranked derivative on Wednesday, and validate with ITC by Friday — all under one project team — compresses traditional 3-month optimization loops into 2-week iterations.

Biacore 8K+

MicroCal PEAQ-ITC

Prometheus NT.48 DSF
Platform specifications are subject to continuous upgrade. Contact our team for instrument availability and project-specific capability assessment.
Closed-Loop Discovery Engine
When Trajectory Meets Thermodynamic Truth
Static structures provide hypotheses. Dynamics tests them against physical reality. Our platform feeds every simulated conformation and predicted free energy back into the design cycle.
Structure Equilibration
AI-predicted or experimental structures are relaxed in explicit solvent, revealing unstable loops and incorrect side-chain rotamers before docking or FEP begins.
→ Feeds into Docking
Dynamic Pocket Discovery
MD trajectories reveal cryptic pockets and allosteric sites invisible in crystal structures, updating virtual screening and pharmacophore protocols with time-resolved accessibility data.
→ Feeds into CADD
FEP-Guided Synthesis
Free energy calculations predict relative affinity changes before compound synthesis, prioritizing modifications with high thermodynamic confidence.
→ Feeds into Wet-lab
Industrial Value:
For Biotechs
Your first FEP campaign calibrates the force field for your target family. Your second campaign inherits that accuracy — predictions sharpen, synthesis cycles drop, and your burn rate is protected by physics.
For Pharma
Every simulated trajectory and free energy prediction is paired with an experimental binding measurement, complete with timestamp, force-field version, and parameter set — fully traceable for regulatory scrutiny and portfolio analytics.
Project Management & Execution
Project Workflow
A standardized, milestone-driven execution system. From system setup to trajectory-derived insight delivery.


01 Setup
- System building: protein structure curation, ligand parameterization, bilayer assembly
- Force-field selection and validation: AMBER, CHARMM, or OPLS with ligand-specific tuning
Deliverable: System setup report with topology, parameter files, and equilibration metrics
02 Simulation
- Production MD: microsecond explicit-solvent trajectories with GROMACS/AMBER GPU acceleration
- Enhanced sampling: REMD, metadynamics, or aMD for rare events
Deliverable: Raw trajectory data and stability diagnostics
03 Analysis
- Trajectory analysis: RMSD, RMSF, cluster analysis, principal component decomposition
- FEP / MM/PBSA free energy calculations and decomposition
Deliverable: Free energy rankings and conformational landscape maps
04 Calibration
- SPR / ITC experimental binding data for FEP calibration
- TSA / DLS thermal stability and aggregation validation
Deliverable: Experimental validation report with ΔG correlation
05 Delivery
- Simulation report with trajectory movies, conformational ensembles, and docking updates
- ADMET risk flags for trajectory-validated chemotypes
Deliverable: Final technical report + data package + Lead Opt transition plan
Sample Requirements
| Sample Type | Specification |
|---|---|
| Target Structure | PDB file or AI-predicted model; preferred resolution < 3 Å for active-site fidelity |
| Ligand Structures | SDF/MOL2 with defined protonation states; or SMILES for automatic parameterization |
| Experimental Data | Known binding affinities (KD, Ki, IC50) for FEP calibration; preferred but not required |
Standard Deliverables
Upon project completion, clients receive comprehensive experimental reports including:
- Production trajectory files (GROMACS/AMBER format) and analysis scripts
- Conformational ensemble clustering and principal component projections
- FEP-derived ΔΔG values with statistical uncertainty estimates
- MM/PBSA per-residue free energy decomposition
- Trajectory-derived docking and virtual screening recommendations
- Follow-up optimization roadmap and Lead Optimization transition plan
Our technical team responds within 24 hours. All inquiries protected under NDA.
Frequently Asked Questions
Case Study
Published Evidence: HT-SuMD Fragment Posing for SARS-CoV-2 Mpro
Reference: Bissaro M, et al. Inspecting the Mechanism of Fragment Hits Binding on SARS-CoV-2 Mpro by Using Supervised Molecular Dynamics (SuMD) Simulations. ChemMedChem. 2021 Jul 6;16(13):2075-2081.
Research Goal: The authors tested whether supervised MD (SuMD) could predict fragment binding modes, and whether classical MD stability metrics could filter false-positive poses.
Published Data:
- Sampling: 23 non-covalent fragments against SARS-CoV-2 Mpro; 2.3 μs classical MD + 6.3 μs HT-SuMD (AMBER14SB/GAFF)
- Accuracy: 48% (11/23) correctly predicted and ranked (RMSDmin < 2 Å); remainder mis-scored or unsampled
- Stability gate: Fragments with RMSFavg < 2.5 Å accounted for 9/11 correct predictions; this threshold retrospectively prioritized all true positives
Industrial Translation:
The authors demonstrated that thermodynamic scoring alone misranks ~50% of fragment poses. Their approach — SuMD posing plus MD stability validation — aligns with our MD-driven screening workflow: trajectories serve as quantitative decision gates, not just visualization. For biotechs, this cuts false-positive synthesis cycles. For pharma, it provides an auditable geometric-dynamic confidence metric alongside FEP rankings.
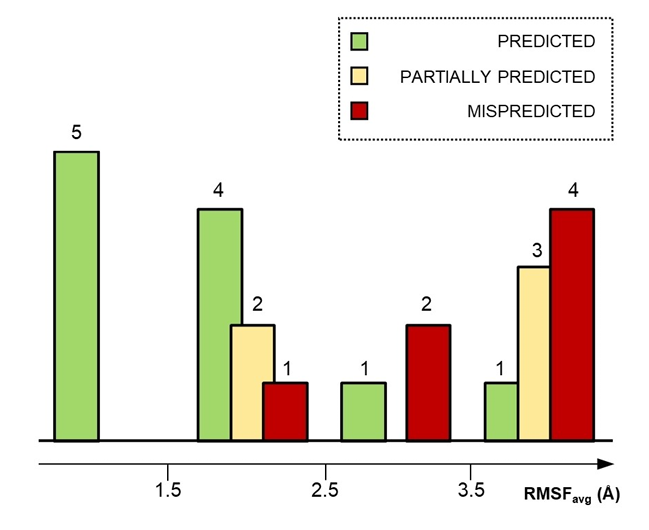
Figure 1. HT-SuMD accuracy vs. fragment pose stability (RMSFavg). Lower RMSF predicts more reliable binding modes. (Bissaro, et al. 2021)