Structure-Guided Medicinal Chemistry and Synthesis
Lead optimization fails when medicinal chemistry, computational design, and structural biology operate in silos. The MagHelix™ platform closes the loop: every R-group modification is FEP-scored, every analog is structurally validated by co-crystal or Cryo-EM, and every dead-end is killed computationally before it consumes synthesis budget. Whether you are a seed-stage biotech without a fume hood or a pharma team pursuing allosteric or covalent programs, we deliver lead candidates with atomic-resolution confidence under one project team and one milestone clock.
Why Structure-Guided Medicinal Chemistry Is the Critical Bridge Between Lead and Candidate
High-throughput screening delivers hits, but hits are not leads, and leads are not candidates. Seed-stage biotechs burn runway on synthesis cycles that should have been killed computationally. Pharma teams lose months coordinating a computational CRO, a medicinal chemistry shop, and a crystallography facility while analogs sit in queues waiting for structural feedback that never arrives.
The MagHelix™ platform eliminates that friction. We integrate generative AI design, GPU-accelerated FEP calculations, and industrial-grade custom synthesis with real-time structural validation. Computed binding poses are validated experimentally in the same project cycle creating a closed loop that refines both prediction accuracy and compound prioritization before your next board meeting.
What Sets the MagHelix™ Platform Apart
Computation-First, Synthesis-Second
Every analog is FEP-scored and ADMET-filtered in silico before a reagent is ordered. AI solubility and synthetic accessibility classifiers eliminate 30–40% of proposed modifications before they consume synthetic budget.
Structural Biology Depth, Not Just SAR Spreadsheets
Because we operate an integrated structural biology pipeline from gene-to-protein to co-crystal soaking priority compounds advance directly into atomic-resolution binding mode determination within days of synthesis completion.
Single-Vendor Accountability
One project team, one data system, one milestone clock. No handoff delays between a computational CRO and a synthesis shop. No lost context when biophysical data passes to medicinal chemists. Your program moves from design to validated candidate without vendor coordination overhead.
Medicinal Chemistry Technology Suite
Structure-Guided Lead Design
Atomic-Resolution Binding Mode Informs Every Modification
Key Features:
- Structure-based docking (Glide, AutoDock-GPU) with induced-fit and covalent docking enabled; designs are anchored to experimental binding poses, not hypothetical models.
- Binding pocket analysis identifies allosteric cryptic sites and druggable sub-pockets for expansion beyond the initial lead chemotype.
- Homology model integration: For targets without crystal structures, AlphaFold2/RoseTTAFold models are refined by MD simulations to generate design-grade structural templates.
Ideal For:
Kinases, proteases, GPCRs, PPI disruptors, and covalent inhibitor programs with available or modelable structural points. What We Offer: For virtual biotechs, this means accessing structure-guided design without maintaining docking software licenses or GPU infrastructure. For pharma teams, it means designs are validated against the same structural biology pipeline that produces your protein ensuring model quality and design fidelity from day one.
AI-Assisted Scaffold Optimization
Generative Chemical Space Exploration with ADMET Guardrails

Key Features:
- Generative AI engine (VAE/GAN/Transformer architectures) explores R-group vectors, scaffold hopping, and ring-system modifications while preserving core pharmacophores.
- Deep learning ADMET panel preemptively flags hERG, CYP450, solubility, and permeability liabilities at the design stage before synthesis commitment.
- Synthetic accessibility scoring ensures every AI-generated analog is rated for route feasibility, eliminating "unmakeable" designs.
Ideal For:
Rapid expansion of chemical space around a validated lead scaffold; escaping patent landscapes; optimizing drug-like properties in parallel with potency. What We Offer: Traditional medicinal chemistry relies on intuition and iterative synthesis. Our closed-loop approach uses AI to explore 10,000+ virtual analogs, ranks them by predicted potency and property profiles, and delivers a focused library of 20–30 high-confidence designs with predicted SAR trajectories. You synthesize only what the models validate.
FEP-Guided Affinity Engineering
Predict ΔΔG Before You Synthesize

Key Features:
- GPU-accelerated FEP/Non-Equilibrium Switching (NES) using GROMACS/AMBER predicts relative binding affinity changes (ΔΔG) for proposed modifications with high correlation to experimental data.
- Thermodynamic decomposition identifies whether affinity gains are enthalpy-driven (H-bond optimization) or entropy-driven (hydrophobic expansion) directing medicinal chemistry strategy.
- MM/PBSA and MM/GBSA scoring provides rapid lead ranking and selectivity assessment against off-target homologs.
Ideal For:
Prioritizing R-group modifications; optimizing linker length in PROTACs/molecular glues; evaluating charge-state changes in polar binding pockets. What We Offer: FEP is the computational difference between guessing and knowing. For seed-stage biotechs, it means 40%+ fewer dead-end synthesis cycles conserving cash and runway. For pharma teams, it means every compound entering the lab has a predicted binding rationale that can be tested structurally, creating an audit-ready design-to-validation chain.
Custom Multi-Step Synthesis
From Milligram to Gram, Route to Reality

Key Features:
- Route scouting and optimization for complex heterocycles, natural product derivatives, chiral centers, and macrocycles.
- Asymmetric synthesis, cross-coupling, and C–H activation capabilities for non-trivial scaffolds that standard CROs decline.
- Scale flexibility: Milligram-scale SAR exploration to gram-scale lead candidate supply for IND-enabling ADMET and formulation studies.
Ideal For:
Multi-step analogs requiring non-commercial starting materials; late-stage functionalization of complex cores; enantiopure chiral synthesis. What We Offer: You receive purified compounds (>95% by HPLC-UV/LC-MS) with full synthetic route documentation, NMR structural verification, and Certificate of Analysis ready for immediate biophysical validation or preclinical profiling. For programs advancing to IND, we provide process chemistry consultation and CMO tech-transfer packages.
Crystallography-Validated Iteration
Atomic Feedback Closes the Design Loop
Key Features:
- Co-crystal soaking and X-ray data collection for each design cycle, typically within 1–2 weeks of compound delivery.
- AI-assisted crystallization screening predicts optimal soaking conditions and cryoprotectants, reducing crystal optimization from months to days.
- Real-time docking protocol updates: Experimentally validated binding modes feed back into induced-fit docking parameters for the next design cycle.
Ideal For:
Structure-driven optimization programs where atomic-resolution binding mode confirmation is required at every iteration. What We Offer: This is where our platform diverges from traditional CROs. When synthesis and crystallography share the same project team, your medicinal chemist receives PDB coordinates and electron density maps not just IC50 spreadsheets within days of compound completion. For covalent inhibitors and allosteric modulators, this structural feedback is the difference between rational optimization and blind iteration.
Platform Instrumentation
| Instrument | Throughput / Sensitivity |
|---|---|
| Waters AutoPurification System | Prep-HPLC; mg-to-g scale purification with mass-triggered fractionation |
| Shimadzu LC-20AP Prep-HPLC | Preparative scale isolation; automated fraction collection |
| Bruker AVANCE NEO 600 MHz | Multi-dimensional NMR; structural verification and stereochemical confirmation |
| Waters Xevo G2-XS QTof | LC-MS/MS; exact mass confirmation and purity assessment |
| Rigaku XtaLAB Synergy | X-ray diffractometer; co-crystal structure determination |
| Thermo Fisher Krios G4 | Cryo-EM; single-particle analysis for large complexes and membrane proteins |
Standardized Workflow
Project Workflow
A standardized, milestone-driven execution system. From lead compound handoff to drug-ready candidate delivery managed by a single project team, tracked in real time, with AI embedded at every decision gate.


01 Strategy & In Silico Design
- Target structure review (co-crystal or homology model) and binding pocket analysis
- AI generative design + FEP affinity prediction for prioritized modifications
- ADMET risk pre-filtering and synthetic accessibility scoring
Deliverable: In silico design report + prioritized compound shortlist (20–30 derivatives)
02 Custom Synthesis
- Route scouting and multi-step organic synthesis execution
- Parallel array synthesis for SAR matrix (if applicable)
- Prep-HPLC/SFC purification to >95% purity
Deliverable: Purified compounds with CoA + synthetic route documentation
03 Analytical QC
- LC-MS/MS identity confirmation and molecular weight verification
- NMR structural verification (¹H, ¹³C, 2D if required)
- HPLC-UV purity and chiral purity assessment
Deliverable: Analytical data package per compound
04 Structural Validation
- Co-crystal soaking and X-ray data collection
- Binding mode determination at atomic resolution
- Cryo-EM validation for large complexes
Deliverable: Structural coordinates (PDB) + electron density maps + binding mode analysis
05 SAR Integration & Next Cycle
- SAR correlation: structure → activity → binding mode
- FEP model recalibration with experimental SPR/ITC data
- ADMET profile update and preclinical risk assessment
- Next-cycle design iteration or Lead Optimization transition plan
Deliverable: Integrated SAR report + validated lead candidate + electronic data package
Sample Requirements
| Sample Type | Specification |
|---|---|
| Starting Lead Compound | ≥10 mg; purity >90%; structural identity confirmed (NMR/HRMS); known IC50/EC50 or binding data |
| Target Protein | For structural validation: ≥95% purity, ≥5 mg/mL, stability data (DSF Tm) recommended |
| Reference Compounds | Known active analogs or competitor molecules for assay benchmarking (if available) |
| Project Background | Target biological function, disease relevance, known SAR constraints, selectivity watch list, off-target liability concerns |
Note: Non-standard formats are welcome. Contact us to discuss your specific requirements.
Standard Deliverables
- In silico design report with prioritized analog shortlist and FEP predictions
- Purified compounds (>95% purity) with CoA, NMR, and LC-MS/MS data
- Co-crystal structures (PDB coordinates + electron density maps) or Cryo-EM validation reports
- Integrated SAR analysis correlating structural, biophysical, and activity data
- ADMET risk assessment and preclinical transition plan
- Final technical report + electronic data package
Frequently Asked Questions
Case Study
Case Study: Accelerating Lead Optimization via Structure-Based Design: A 10,000-Fold Potency Leap in TBK1 Inhibitor Development
Goal: Transform a micromolar TBK1 kinase hit into a picomolar lead through a computation-first, structure-guided optimization paradigm, establishing a benchmark for efficient scaffold morphing and MD-driven compound prioritization.
Key Data:
- Computation-first funnel: 5,000+ virtual analogs screened by MD/MM-PBSA → only 14 compounds synthesized.
- Potency leap: Lead compound A1 achieved IC50 = 775 pM, a >10,000-fold improvement over the initial hit.
- Selectivity: High target precision confirmed across an 82-kinase counter-screening panel.
- Hit rate: 5 of 14 synthesized compounds reached nanomolar-level potency (~36% success rate).
Why it matters: In traditional medicinal chemistry, teams often synthesize hundreds of analogs to achieve a single viable lead, with success rates below 5%. This peer-reviewed case demonstrates that computational pre-validation inverts that equation: by anchoring design to MD-simulated binding poses and MM-PBSA energy rankings, the research team synthesized only 14 compounds yet achieved a 10,000-fold potency improvement and a 36% nanomolar hit rate. For drug developers, this establishes a published precedent that structure-guided, computation-first workflows can compress lead optimization timelines by months while dramatically reducing synthetic dead-ends.
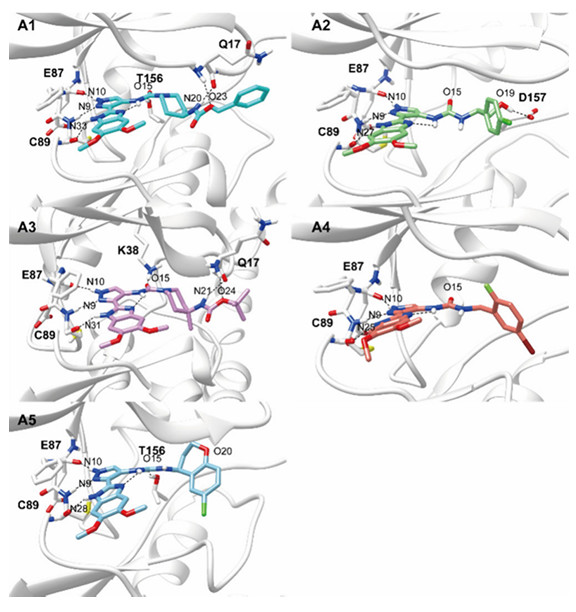
Figure 1. Representative binding poses of designed compounds with TBK1 during MD simulation. (Sun W, et al., 2025)
Reference
- Sun W, Xie Y, Xia Q, et al. Structure-Based Optimization of TBK1 Inhibitors. ACS Med Chem Lett. 2025 Mar 31;16(4):611-616.
Our technical team responds within 24 hours. All inquiries protected under NDA.