Lead Optimization
Most lead optimization programs die not because chemists lack skill, but because the strategy lacks structural certainty. We replace iterative synthesis loops with a structure-first, AI-guided approach: every chemical modification is validated by co-crystal data, every liability is flagged by predictive ADMET models before synthesis, and every lead candidate exits with a comprehensive optimization dossier — potency, selectivity, ADMET, and structural rationale ready for handoff to your development team.
Creative Biostructure at a Glance
Why Partner With Us
The bridge from lead to development-ready candidate is where most discovery programs collapse. Conventional "synthesize-and-test" cycles burn cash and time because they lack real-time structural feedback. We built this platform to ensure every chemical modification is computationally rationalized and structurally validated before it reaches the lab — compressing 18-month traditional timelines into focused, milestone-driven campaigns.
Your CapEx is in chemistry partnerships and disease biology. Ours is in structural biology and computational optimization.
| Stage | What We Deliver | What You Don't Need to Build |
|---|---|---|
| Structure-Guided Design | Co-crystal structures / Cryo-EM maps guiding R-group modifications; FEP-scored affinity predictions | Structural biology team, synchrotron access |
| AI-ADMET Filtering | Transformer models flagging hERG, CYP450, solubility, permeability liabilities before synthesis | Computational toxicology infrastructure |
| Synthesis Coordination | Focused library design (20–30 derivatives); route feasibility scoring; CRO coordination or internal handoff | Medicinal chemistry FTE headcount |
| Data Handoff | SAR report, structural passport, ADMET risk assessment, development transition documentation | — |
Production-Ready Deliverables: Every optimized lead ships with co-crystal coordinates, thermodynamic binding data, FEP-derived affinity trajectories, and a complete development dossier — ready for your internal development team or preclinical partner.
- ✓ Milestone-based pricing aligned with your fundraising cycles
- ✓ No full-time medchem headcount required — structure-guided design reduces synthesis cycles by 40%+
80% of projects stall in lead optimization. We prevent late-stage attrition by front-loading structural and ADMET certainty.
Structure-first optimization
Every SAR decision is anchored to atomic-resolution co-crystal structures or Cryo-EM maps — not intuition. This eliminates the "molecular bloat" that kills developability.
AI-ADMET risk flagging
Deep learning models preemptively identify metabolic soft spots, solubility cliffs, and hERG liabilities before they become expensive late-stage surprises.
Optimization data integrity
All structural, biophysical, and ADMET data generated under GLP-ready protocols with full audit trails — formatted for seamless handoff to internal development teams or CRO partners.
Core Service Modules
Service Module At-a-Glance
| Service | Core Capability | Structural + Computational Integration | Typical Timeline |
|---|---|---|---|
| Structure-Guided Medicinal Chemistry | Lead synthesis, complex intermediate development, chiral synthesis; scaffold hopping | SBDD platform with binding free energy calculations; scaffold hopping algorithms for IP navigation | 8–16 weeks per design cycle |
| SAR Analysis | Systematic substituent exploration; quantitative activity gradient mapping | FEP predicts precise modification contributions; molecular docking guides substitution site selection | 4–8 weeks per iteration |
| Property-Based Optimization (PBO) | Solubility, permeability, metabolic stability improvement | AI-ADMET prediction pre-synthesis; QSAR models for property correlation | 6–12 weeks |
Structure-Guided Medicinal Chemistry & Synthesis
Every Synthesis Decision Anchored to Atomic-Resolution Data
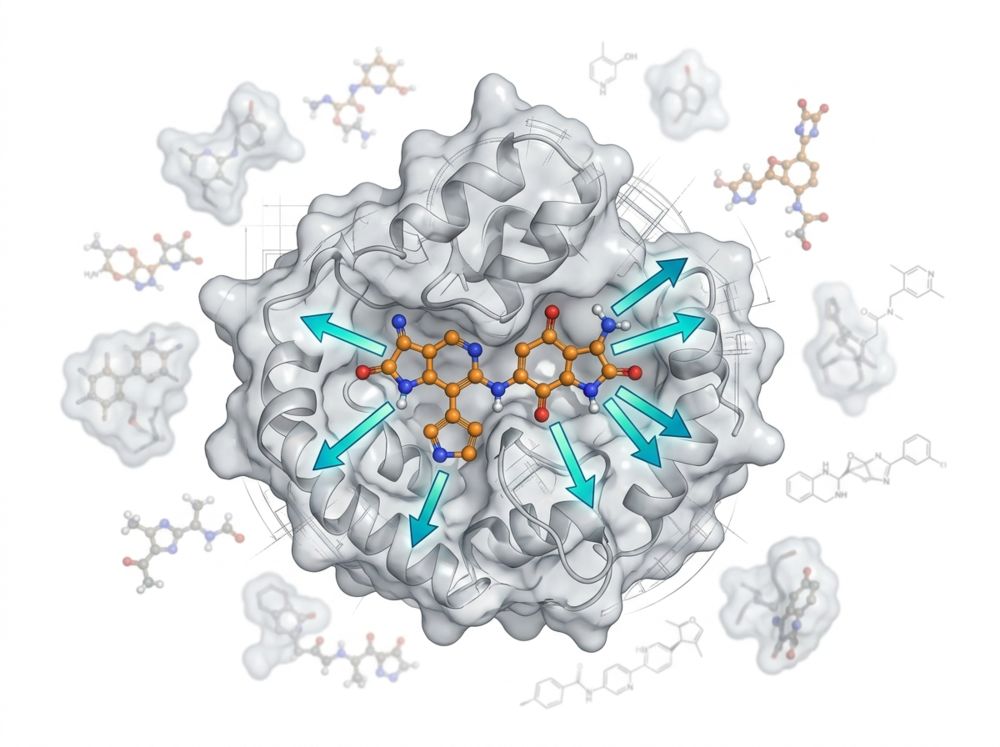
Key Features of Our Medicinal Chemistry Services:
- Structure-First Design Cycles — Co-crystal structures and Cryo-EM maps guide R-group placement, not intuition. We visualize exactly how each modification impacts the binding pocket before ordering synthesis.
- Scaffold Hopping & IP Navigation — AI generative models and shape-based screening identify novel, patent-free core scaffolds that maintain key interactions while circumventing competitor IP.
- Flexible Synthesis Models — Coordinated CRO synthesis or direct FTE collaboration; route feasibility scoring ensures every designed compound is makeable.
What We Offer:
For biotechs without internal medicinal chemistry, we operate as your virtual drug design and synthesis coordination team. For pharma, we provide a structure-guided satellite team that accelerates your internal pipeline without adding permanent headcount.
Explore Medicinal Chemistry →SAR Analysis
Quantitative Structure-Activity Relationships Driven by Physics
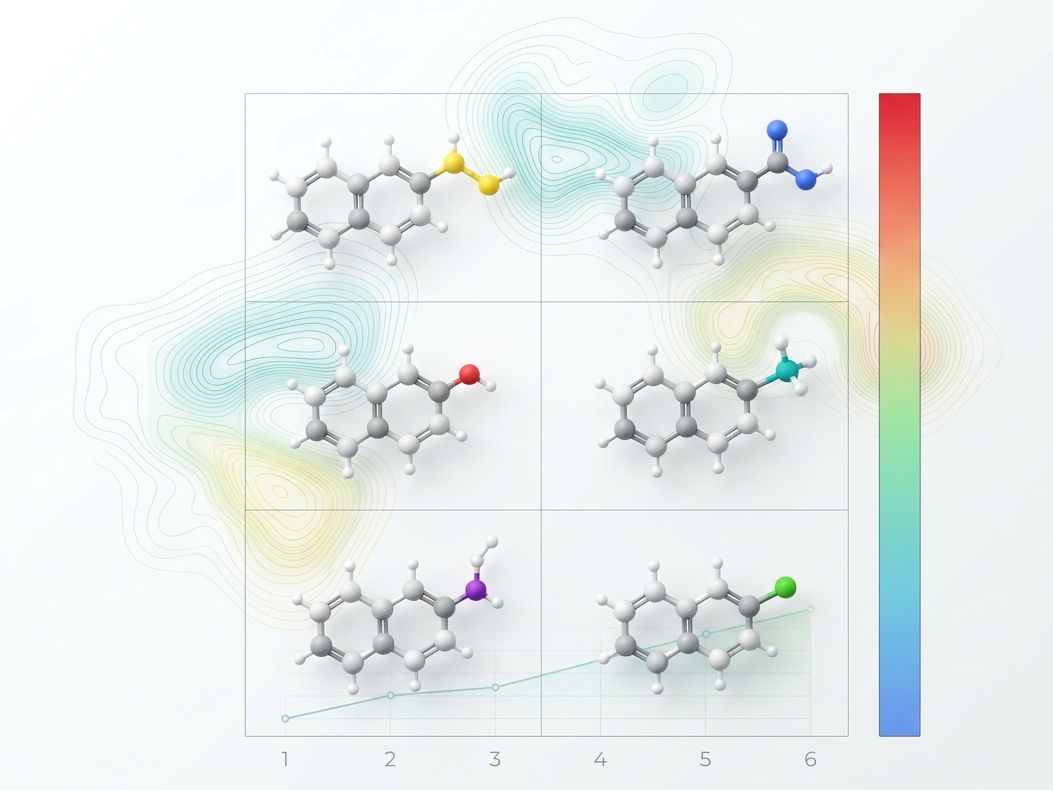
Key Features of Our SAR Services:
- Systematic Substituent Mapping — Grid-based exploration of electronic, steric, and hydrophobic effects on target activity and selectivity.
- FEP-Guided Prioritization — Free Energy Perturbation predicts the precise thermodynamic contribution of each modification, directing synthesis toward high-confidence changes.
- Multi-Dimensional Optimization — Simultaneous tracking of potency, selectivity, solubility, and metabolic stability across analog series.
What We Offer:
Traditional SAR relies on synthesizing 50+ analogs to find a trend. Our FEP-assisted approach identifies the 10–15 most informative modifications first, reducing analog counts and accelerating convergence on optimal leads.
Explore SAR Analysis →Property-Based Optimization (PBO)
ADMET-by-Design, Not ADMET-by-Accident
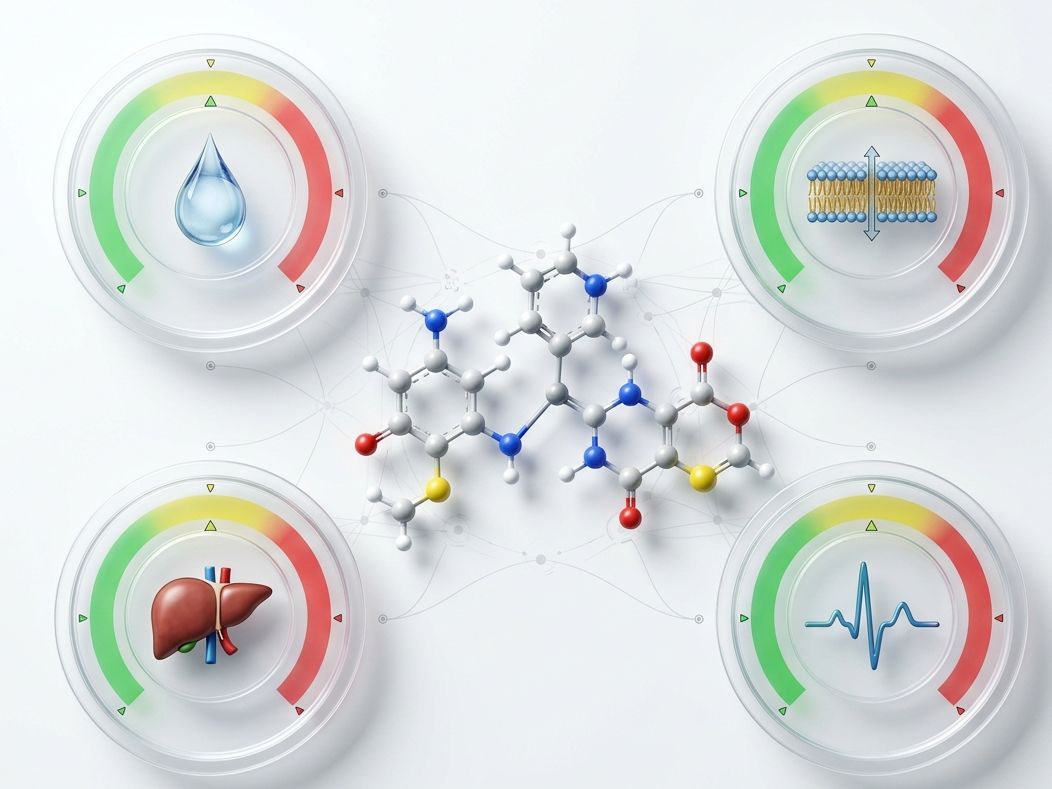
Key Features of Our PBO Services:
- AI-ADMET Pre-Filtering — Transformer-architecture models assess hERG, CYP450 inhibition, microsomal stability, P-gp substrate liability, and BBB permeability before a compound is synthesized.
- Solubility & Permeability Engineering — Structure-based modifications to introduce polarity vectors or disrupt crystalline packing without sacrificing target binding.
- Metabolic Soft Spot Identification — MD simulations and QSAR models flag metabolic liabilities; deuteration or bioisostere strategies proposed pre-synthesis.
What We Offer:
Selectivity failures and ADMET surprises kill programs in development. Our front-loaded computational profiling ensures your lead candidate enters the next phase with a clean liability profile — not a ticking time bomb.
Explore Property-Based Optimization →Technology Platform
Integrated Lead Optimization Infrastructure: Structure + Computation + Synthesis
Our platform spans atomic-resolution structural biology, physics-based affinity prediction, and AI-driven property modeling — enabling lead evolution without the delays of coordinating a crystallography facility, a CRO, and a computational group.
Dry Lab — Computational Platform
Powered by our MagHelix™ CADD Platform and MagHelix™ AIDD Platform
| Instrument / Infrastructure | Role in Lead Optimization |
|---|---|
| NVIDIA DGX A100 | GPU-accelerated cluster for GROMACS/AMBER microsecond-scale MD and generative AI model training |
| MagHelix™ CADD Compute Cluster | Dedicated high-performance computing infrastructure integrating FEP, molecular docking, and ADMET prediction for real-time SAR decision support |
| Elastic Cloud Screening Infrastructure | Scalable compute architecture supporting 10^8-scale virtual screening campaigns and AI-driven focused library generation |

NVIDIA DGX A100

MagHelix™ CADD Compute Cluster

Elastic Cloud Screening Infrastructure
Wet Lab — Structural & Biophysical Validation
Powered by our MagHelix™ Structural Biology and SBDD Platform
| Instrument / Model | Role in Lead Optimization |
|---|---|
| Rigaku Synergy-R | X-ray crystallography with automated crystal screening robots for rapid lead compound co-crystal structure determination |
| Thermo Scientific Krios G4 | Cryo-EM for membrane protein–lead complex structures when crystallization fails |
| Biacore 8K+ | SPR system for real-time binding kinetics (kon/koff) tracking SAR progression across analog series |
Platform Edge: The ability to generate co-crystal structures within days of synthesizing a new analog — then feed those coordinates back into FEP calculations for the next design cycle — compresses traditional 3-month SAR loops into 2-week iterations.

XtaLAB Synergy-R

Thermo Fisher Krios G4

Biacore 8K+
Closed-Loop Discovery Engine
When Structural Validation Meets Computational Design
Traditional medicinal chemistry separates design from structural feedback — chemists synthesize, then wait weeks for crystallography results. Our platform operates as a closed-loop system: every co-crystal structure feeds back into our AI and FEP models in real time.
Structure-Guided SAR Refinement
Co-crystal structures of lead analogs feed back into docking and scoring functions, refining induced-fit parameters and eliminating incorrect design hypotheses before the next synthesis cycle.
Co-crystal → Docking/Scoring
FEP-Calibrated Affinity Prediction
Experimental IC50/SPR binding data calibrates target-class-specific FEP parameters, improving ΔΔG prediction accuracy for subsequent analog designs.
Biophysics → FEP Models
ADMET-Aware Generative Design
In vitro ADMET liabilities (hERG, CYP450, solubility) train multi-parameter optimization functions in our generative AI, directing synthesis toward developable scaffolds.
ADMET Data → Generative AI
Thermal Stability Feedback
NanoDSF/DSC thermal unfolding data identifies destabilizing modifications, informing computational stability predictions and guiding away from aggregation-prone chemotypes.
Thermal Data → MD Stability
Industrial Value:
For biotechs
Your lead program calibrates our models for your next target. Structural data from your current campaign becomes training data for the next — a compounding learning partnership.
For pharma
Every structural determination is linked to a computational prediction with project ID, timestamp, and model version — fully audit-ready for development handoff.
Project Management & Execution
Project Workflow
A standardized, milestone-driven execution system. From lead assessment to development-ready data package.


01 Lead Assessment
- Consolidate Hit-to-Lead data: co-crystal structures, binding kinetics, preliminary SAR
- Define optimization objectives: potency, selectivity, solubility, metabolic stability
Deliverable: Optimization proposal with Gantt-chart milestones and risk matrix
02 Structure-Guided Design
- FEP scoring of proposed modifications; AI-ADMET liability flagging
- Scaffold hopping or R-group exploration as needed
Deliverable: 20–30 derivative designs with predicted profiles and synthetic routes
03 Synthesis & Testing
- Focused library synthesis; IC50/EC50 determination; SPR binding kinetics
Deliverable: Experimental potency and selectivity dataset
04 ADMET & Selectivity
- AI-ADMET validation; hERG, CYP450, microsomal stability, permeability screening
- Safety panel counter-screening; homolog protein selectivity
Deliverable: ADMET risk assessment and selectivity profile
05 Lead Candidate Package
- Comprehensive SAR report with structural rationale
- Co-crystal structures / Cryo-EM coordinates
- Thermodynamic binding data (ITC, SPR)
- Development handoff documentation and follow-up optimization roadmap
Deliverable: Final technical report + electronic data package + lead candidate dossier
Sample Requirements
To ensure a precise starting point for optimization experiments, we recommend clients provide:
| Sample Type | Specification |
|---|---|
| Lead Compounds | Well-defined candidate molecules (SDF format), purity ≥95% recommended |
| Activity Data | Previous experimental IC₅₀, EC₅₀, or binding kinetic parameters |
| Target Information | High-resolution protein structural data or high-purity protein samples (for subsequent complex structure determination) |
Standard Deliverables
- Optimized Molecules: IP-protected optimized lead compounds with detailed synthetic routes
- Comprehensive SAR Report: Including activity testing data for all analogs, quantitative structure-activity relationship analysis, and relevant curve maps
- Computational & Structural Data: Protein-lead compound complex structure coordinate files and binding mode prediction reports
Our technical team responds within 24 hours. All inquiries protected under NDA.
Frequently Asked Questions
Case Study
Structure-Guided SAR: 2 Å Co-Crystal of a Protein–Nucleic Acid Complex
Goal: Resolve atomic-resolution structure of a protein–DNA/RNA complex to establish the structural foundation for structure-based lead optimization — a prerequisite for rational R-group design on nucleic-acid-binding small molecules.
Key Data:
- Target: XYZ protein–DNA/RNA complex (customer program, produced in-house)
- Resolution: 2 Å; Space group: I222 (a = 80.330 Å, b = 82.423 Å, c = 126.061 Å)
- Phasing: Molecular replacement using PDB 4MH8 homology model, split-domain strategy (T24–I261 + Q263–T482) with nucleic acid component
- Optimization trajectory: Initial screening (8–10 Å) → cryo-buffer optimization (0.05 M CaCl₂, 0.05 M NaAc, pH 5.0, 28% MPD, 10% glycerol) → final 2 Å
Why it matters: Nucleic acid–protein complexes are among the most challenging structural targets in drug discovery — and among the most valuable, as they represent a vast untapped chemical space beyond traditional protein pockets. This case demonstrates our end-to-end structural enablement for non-traditional targets: from protein/nucleic acid production and complex reconstitution to reproducible crystallization and synchrotron data collection. For biotechs pursuing novel target classes, this means accessing synchrotron-grade structural biology without building an internal crystallization facility. For pharma outsourcing teams, this means a single accountability chain that delivers the atomic-resolution blueprint required for structure-guided lead optimization — transforming what would otherwise be a "blind" medicinal chemistry campaign into a rational, electron-density-driven SAR process.
Figure 1. Optimized XYZ–DNA/RNA crystals after 8-week incubation (0.2 M CaCl₂, pH 5.0, 40% MPD).

Figure 2. X-ray diffraction to 2 Å resolution (SSRF BL18U1, λ = 0.9791 Å).
Figure 3. Dual-domain molecular replacement solution (PDB 4MH8) for the XYZ–DNA/RNA complex.
ITC Thermodynamic Profiling: Quantifying Lead–Target Binding Mechanism
Goal: Decompose lead–target binding into enthalpic and entropic contributions to guide rational SAR decisions.
Key Data:
- Association constant (K): 1.20 × 10⁵ M⁻¹ — strong binding affinity
- Enthalpy (ΔH): −3.44 × 10⁵ cal/mol — enthalpy-driven, favorable H-bond / vdW contacts
- Entropy (ΔS): −1.13 × 10³ cal/mol/deg — negative, indicating ordered stable complex
- Stoichiometry (N): 0.106 — sub-stoichiometric, flags need for protein/buffer condition refinement
Why it matters: IC50 tells you that a compound binds. ITC tells you how to optimize it. The strongly negative ΔH reveals this lead is enthalpy-driven — directing medicinal chemists to invest in polar R-group modifications (H-bond networks) rather than hydrophobic expansion. For biotechs, this means informed SAR bets without an ITC facility. For pharma, this means thermodynamic data packages that satisfy development scrutiny.
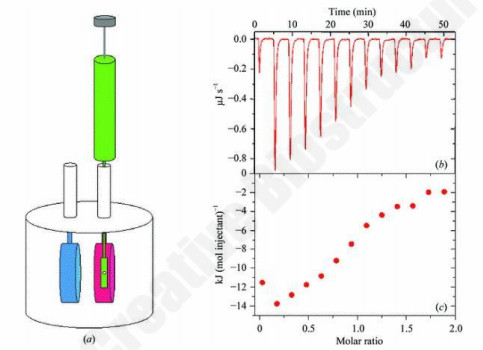
Figure 1. ITC platform: Label-free thermodynamic characterization.
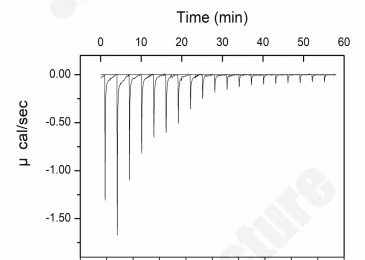
Figure 2. Raw thermogram: Exothermic binding signature.
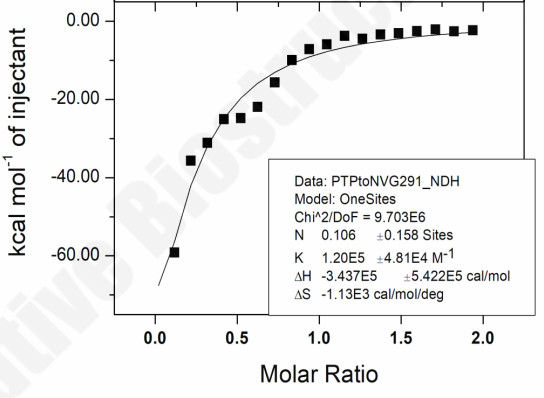
Figure 3. Thermodynamic profile: Enthalpy-driven binding mechanism.