Computational Biology & AI: Decoding Biological Complexity with Atomic-Level Precision
Integrating Generative AI, Physics-Based Simulations, and High-Performance Computing to transform raw biological sequences into actionable structural insights. Our platform empowers researchers to navigate "Chemical Dark Matter" and solve the most challenging structural puzzles in modern drug discovery.
Overcoming the Limits of Traditional Modeling
The Challenges in Modern Computational R&D
Modern drug discovery is increasingly focused on high-complexity targets such as GPCRs, ion channels, and Protein-Protein Interactions (PPIs). Traditional computational methods often face significant technical bottlenecks:
* Sampling Efficiency: Difficulty in capturing the full conformational landscape of flexible proteins or disordered regions.
* Scoring Accuracy: High attrition rates due to the lack of chemical accuracy in predicting binding free energy and SAR.
* Structural Uncertainty: Traditional linear screening often fails to predict ADMET and binding stability early in the pipeline.
Our Strategic Response: An Integrated Computational Framework
To address these gaps, we implement a Physics-ML Hybrid Framework that spans the entire spectrum of computational biology:
* Beyond Static Structures: Utilizing AlphaFold-guided conformational analysis and Enhanced Sampling Molecular Dynamics (MD) to reveal cryptic binding pockets that traditional screening misses.
* Predictive Accuracy: Integrating FEP (Free Energy Perturbation) to achieve experimental-grade affinity predictions in silico, minimizing redundant wet-lab iterations.
* Generative Innovation: Leveraging RFdiffusion and ProteinMPNN for de novo design, creating high-affinity binders for surfaces that lack traditional small-molecule pockets.
* Antibody & Protein Engineering: Applying deep learning for Antibody Humanization, Antibody-Antigen Interaction Modeling, and Protein Stability Optimization.
Reduction
Success Rate
Simulations
Countries
Integrated Computational Solutions for Molecular Innovation
From de novo design to atomic-level affinity quantification, our computational biology services provide the predictive power needed to bypass traditional R&D bottlenecks and accelerate your drug discovery journey.
AI Focus: AlphaFold-3, IgFold/DeepAb, and Co-evolutionary Analysis algorithms.
High-precision modeling covering AlphaFold prediction, homology modeling, and ab initio methods. We provide comprehensive structure refinement, pocket druggability analysis, and specialized complex/antibody structure prediction.
AI Focus: Induced-fit docking, Pharmacophore modeling, and AI-driven virtual screening.
Advanced solutions for molecular docking (PPI, covalent, and ligand), pharmacophore screening, and large-scale virtual screening services to optimize drug design and library analysis for diverse targets.
AI Focus: FEP+, MM/PBSA, and Enhanced Sampling (REMD) techniques.
Dynamic insights via all-atom, membrane, and nucleic acid MD simulations, featuring high-precision binding free energy calculations and coarse-grained modeling to explore long-term biological processes.
AI Focus: Deep learning-based humanization and affinity maturation platforms.
Precision engineering of therapeutic biologics, including in silico antibody humanization, affinity maturation, and predictive profiling of aggregation, solubility, and epitope/paratope mapping.
AI Focus: Graph Neural Networks (GNN) and AI-based QSAR analysis.
Rapid de-risking of candidates through AI-based toxicity prediction (hERG, Ames), metabolic stability assessment, and QSAR analysis to ensure optimal drug-likeness and safety profiles.
Service Selection Guide - Accelerating Your R&D Pipeline
This guide is designed to help R&D scientists select the optimal computational tools based on project maturity and specific technical hurdles. From early-stage target validation to high-precision lead optimization, we provide the exact level of resolution required.
| R&D Stage | Specific Scientific Challenge | Recommended Technical Service | Key Algorithm/Methodology |
|---|---|---|---|
| Target Discovery | Lack of experimental crystal structures for novel or "undruggable" targets. | AI-Based Structure Prediction | AlphaFold-3, Ab Initio Modeling, Pocket Analysis |
| Hit Identification | Screening ultra-large chemical libraries for novel scaffolds against a defined pocket. | CADD & Virtual Screening | Induced-fit Docking, Pharmacophore Modeling |
| Antibody Design | Reducing immunogenicity or increasing affinity of a therapeutic lead candidate. | AI for Antibody & Biologics | Deep Learning-based Humanization, CDR Looping |
| Lead Optimization | Precisely ranking a congeneric series of compounds to minimize wet-lab synthesis. | Molecular Dynamics (MD) Simulations | FEP+ (Free Energy Perturbation), MM/PBSA |
| Candidate Selection | Predicting human metabolic stability and safety profiles (e.g., hERG) before in vivo trials. | ADMET Prediction & Modeling | GNN-based Toxicity Prediction, QSAR Analysis |
Workflow & Methodology - From Algorithm to Insight
Our computational pipeline provides a seamless transition from raw sequence data to validated leads by integrating state-of-the-art AI with physics-based refinements.
Project Assessment & Feasibility Study
We evaluate target druggability using AI-driven cryptic pocket identification and structural analysis to define the optimal R&D roadmap.
Multiscale Simulation & Design
We execute core technical tasks using AlphaFold-3, generative design, or virtual screening, ensuring candidates capture the most energetically favorable biological states.
High-Precision Refinement & Scoring
We apply FEP+ and Enhanced Sampling MD to achieve "chemical accuracy" in affinity predictions, enabling precise rank-ordering of lead molecules.
Actionable Deliverables & Insights
We provide comprehensive technical reports featuring high-resolution 3D coordinates (PDB/SDF), thermodynamic profiling, and strategic guidance for subsequent in vitro assay development.
Sample Requirements & Deliverables
To ensure the highest predictive accuracy, we maintain a transparent process from initial data submission to the final delivery of actionable research insights.
Sample Requirements
- •
Target Sequence & Structural Data We require the protein primary sequence (FASTA) or high-resolution crystal structures (PDB) to initiate homology modeling and binding site analysis.
- •
Chemical Library & Ligand Information For docking and screening, please provide ligand structures in SDF/MOL2 format, including known activity data to calibrate our AI-scoring algorithms.
- •
Specific Research Objectives Clearly defined project goals, such as potency targets, selectivity requirements, or ADMET constraints, help us tailor the computational R&D roadmap.
Deliverables
- •
High-Resolution Structural PackagesWe deliver optimized PDB/SDF files containing atomic coordinates, refined binding poses, and interaction maps for direct integration into your molecular viewers.
- •
Quantitative Thermodynamic DataComprehensive datasets featuring predicted binding affinity, Kd values, and stability scores to enable high-confidence candidate rank-ordering.
- •
Actionable Technical ReportsIn-depth documentation of our methodology, AI-driven SAR insights, and strategic recommendations to guide your subsequent in vitro validation and synthesis.
Case Study
Case: Unified Modeling of Biomolecular Complexes and Ligand Interactions via AlphaFold-3
Background: Accurate prediction of how proteins interact with ligands—including small molecules, nucleic acids, and ions—is a cornerstone of modern drug discovery. This study demonstrates how AlphaFold-3 utilizes a novel diffusion-based architecture to achieve unprecedented accuracy in modeling complex biomolecular systems within a single unified framework.
Key Technical Milestones:
- Integrated Biomolecular Prediction: Unlike previous iterations, AlphaFold-3 enables the joint structure prediction of proteins, nucleic acids (DNA/RNA), small molecules, ions, and modified residues. This "all-in-one" modeling allows for a holistic view of the drug-target environment.
- Superior Ligand-Binding Accuracy: AlphaFold-3 significantly outperforms traditional docking tools and specialized predictors in identifying protein-ligand interactions. Its "chemical accuracy" in positioning small molecules within binding pockets provides high-confidence starting points for lead optimization.
- Enhanced Antibody-Antigen Precision: The model demonstrates substantially higher accuracy in predicting antibody-antigen interfaces compared to AlphaFold-Multimer v2.3. This capability is critical for engineering high-affinity biologics and understanding immune recognition.
- Streamlined Structure-Based Design: By providing high-accuracy modeling across the entire biomolecular space, AlphaFold-3 reduces the need for multiple specialized tools. This streamlines the in silico workflow, allowing researchers to explore novel therapeutic modalities like molecular glues and PROTACs with higher success rates.
Conclusion: AlphaFold-3 represents a paradigm shift in computational biology. By leveraging its ability to predict complex interactions with atomic-level precision, researchers get a transformative R&D edge, drastically reducing the time and cost associated with early-stage drug discovery and experimental validation.
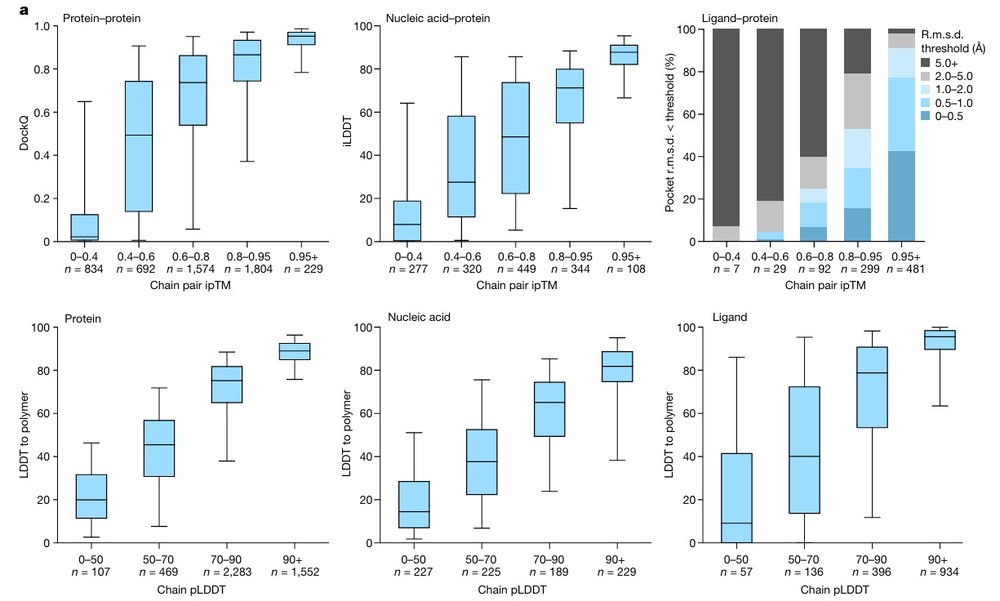
Figure 1. Crystal structures and FP titrations of GCG binders showing structural similarity and enhanced binding after partial diffusion (Vázquez Torres S, et al., 2024).
Why Trust Our Lead Discovery?
Multidisciplinary Ph.D. Team
Our core scientific team consists of Ph.D. experts specialized in computational biology, medicinal chemistry, and structural biology, bringing an average of 10+ years of industrial R&D experience to every project.
State-of-the-Art AI Architecture
We integrate the latest industry-leading algorithms, including AlphaFold-3 and RFdiffusion, with our proprietary MagHelix™ platforms to provide unparalleled predictive accuracy for complex biological systems.
High-Performance Computing Infrastructure
Our specialized GPU clusters and optimized parallel processing workflows ensure rapid turnaround times for large-scale virtual screening and microsecond-scale MD simulations without compromising on data resolution.
Integrated "Dry-to-Wet" Validation
Unlike pure computational firms, we provide seamless transition to wet-lab validation, utilizing X-ray crystallography and SPR to experimentally confirm in silico leads and de-risk your clinical pipeline.

Frequently Asked Questions
For any inquiries, our drug discovery experts are ready to help you get technical support to accelerate your hit-to-lead transition and maximize the success rate of your preclinical candidates.