Obtaining structural information about individual target proteins or complexes of target proteins with other molecules (e.g., proteins, nucleic acids, and small molecules) can provide insight into the structure-function relationship of the target protein and the underlying mechanism of interaction, which is significant for drug discovery and development. However, it is relatively difficult and time-consuming to determine the three-dimensional (3D) structure of biological macromolecules through experimental approaches such as X-ray crystallography, cryo-electron microscopy, or NMR spectroscopy. As a structure-based drug discovery CRO, Creative Biostructure not only provides experimental techniques to determine the structure, but also utilizes high-performance computing tools to predict the structural model of the target molecule, collect information about dynamic properties, and predict binding patterns and affinity between target proteins and other interacting molecules.
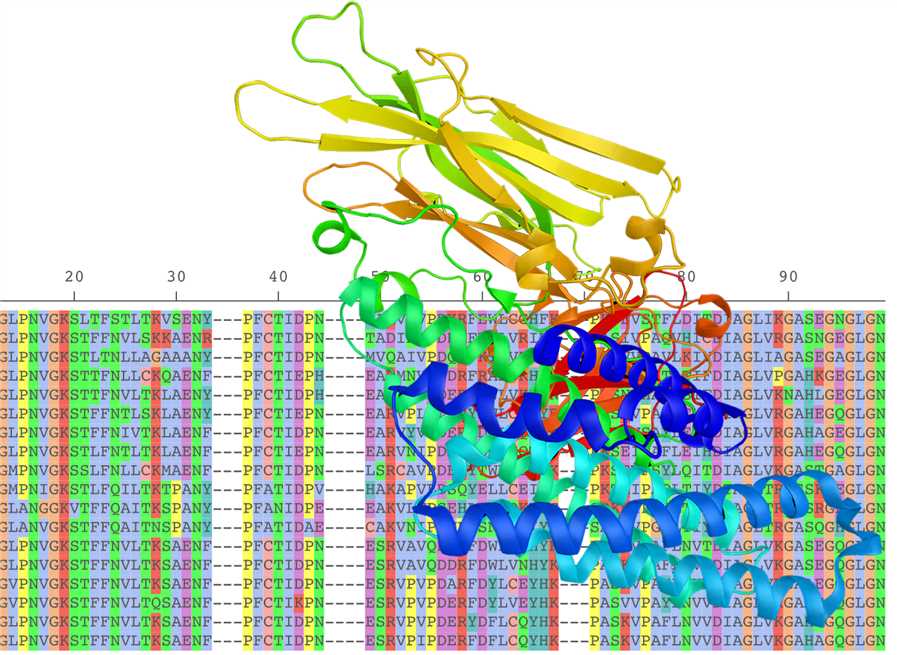
Creative Biostructure can use genetic data or amino acid sequences provided by customers to build target models through computational simulation. Although the structure generated by protein modeling is low-resolution and subject to considerable uncertainty, it contains sufficient information about the spatial arrangement of crucial residues in the target protein and can be used to guide the design of experiments such as site-directed mutagenesis. This predictive structure is also of important reference value for structure-based drug discovery and drug design, especially for some cases where the 3D structure determination of the target encounters bottlenecks due to experimental technical difficulties. Creative Biostructure has extensive expertise and rich experience in generating structural models of various types of proteins and complexes. We mainly adopt two strategies for building target models, namely homology modeling and ab initio modeling, and we can also customize and perform target modeling according to customer requirements. We ensure the reliability of the resulting model for downstream applications, such as virtual screening and structure-based drug design guidance, through rigorous parameter setting and iterative computational validation and simulation.
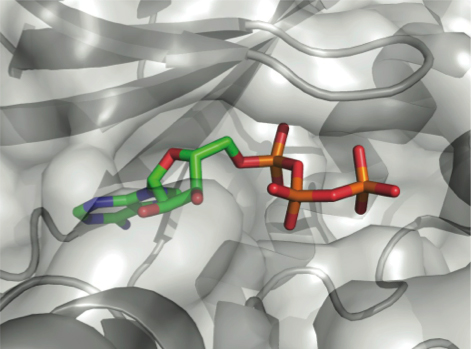
Modeling the interactions between target proteins and other molecules using appropriate computational techniques can help understand the mechanisms of such interactions and provide crucial clues for drug discovery. Computational docking is a significant method to predict the 3D structures of these interacting partners. Creative Biostructure can perform various types of docking and has successfully executed protein-protein docking, antibody-antigen docking, protein-peptide docking, protein-nucleic acid docking, protein-lipid docking, protein-carbohydrate docking and protein-ligand docking for customers in the academia and the pharmaceutical industry. As a drug discovery CRO, we emphasize the ability in protein-ligand computational docking. We can also seamlessly collaborate the computational docking procedure with upstream and downstream computational modeling protocols, which will create more opportunities for structure-based drug design and drug discovery.
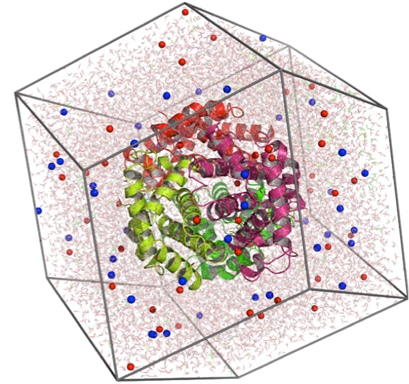
In addition to constructing static target models, we can also use the in-silico simulation technique to offer atomic-level information on the conformational changes and binding thermodynamics of target molecules over time or under predetermined physiological conditions, thereby accelerating drug discovery. Molecular dynamics simulation is a powerful tool for studying condensed matter systems. In this method, atoms and molecules can interact within a given time, thus providing a view of the dynamic evolution of the system. Creative Biostructure utilizes a variety of molecular dynamics software with comprehensive and effective molecular dynamics codes to collect information about the dynamic properties of target molecules and docked complexes (e.g., target-ligand).
Using high-performance computing equipment and sophisticated molecular dynamics simulation algorithms, we are able to simulate and study the dynamics of entire soluble proteins, membrane-embedded proteins, and large biomolecular complexes (e.g., ribosomes, nucleosomes). Moreover, molecular dynamics simulation is an important part of computer-aided drug discovery (CADD), and it can be applied to identify hidden or allosteric binding sites, assist virtual screening, and directly predict the binding energy of ligands.
