Ligand-Based Virtual Screening (LBVS)
Ligand-based virtual screening (LBVS) is a good option for hit identification if the three-dimensional (3D) structure of the target is unknown or if it is challenging to perform virtual drug screening through structure-based methods. The LBVS strategy is based on the assumption that molecules with similar structures (e.g., chemical structures, pharmacophores, molecular fields) tend to have similar properties and functions. Methods of screening potential hits based on physical, chemical, and thermodynamic properties have proven to be reliable, and techniques such as substructure mining and fingerprint searches of LBVS are less time-consuming than the molecular docking technique of structure-based virtual screening (SBVS) strategy. Creative Biostructure provides LBVS solutions to support drug discovery projects. Generally, we first generate fingerprints with high dimension of features to describe the chemical characteristics of ligands and then apply similarity searching or machine learning approaches for screening in high-dimensional data.
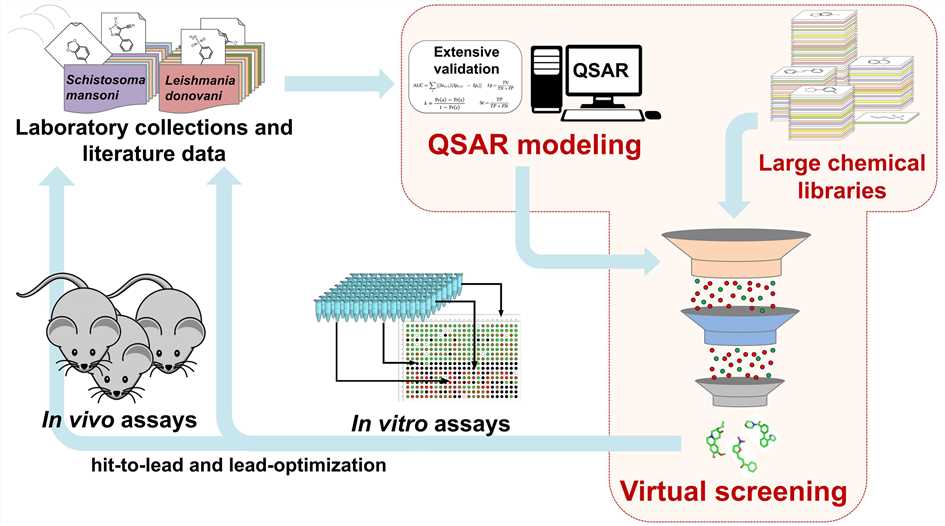 Figure 1. QSAR-based virtual screening. (Neves B J.; et al. 2018)
Figure 1. QSAR-based virtual screening. (Neves B J.; et al. 2018)
Our LBVS approaches include but are not limited to:
- Similarity searching
This is the most rapid and straightforward LBVS method to search for compounds that are physiochemically similar to the query molecule. Similarity can be measured by combining molecular descriptors (physicochemical properties, 2D properties, and 3D properties) with similarity coefficient and weighting function. We utilize data fusion and machine learning to improve the effectiveness of similarity searching.
We can conduct pharmacophore study on a series of known active compounds, and obtain information about some groups that play a key role in the activity of compounds utilizing conformational analysis, molecular superposition and other methods, thereby generating pharmacophore models, and select the models to perform virtual screening with compatible compound databases.
We are able to build a QSAR model using knowledge of known active and known inactive compounds, and then use the model to predict the biological properties of new compounds, that is, whether they belong to active or inactive nodes. In QSAR-based virtual screening, we adopt data curation methods and machine learning algorithms capable of deal with a large number of compounds.
Advantages of our Ligand-based Virtual Screening services:
- Diverse compound databases for screening
- Equipped with servers and screening algorithms that support high-throughput and large-scale LBVS
- Rich experience in the development of pharmacophore and QSAR models involved in LBVS
- Our drug discovery scientists have the ability to prioritize the most promising hits
- Considering cost-effectiveness
In the era of big data, Creative Biostructure continues to increase more computational power to support the screening and evaluation of more compounds. We improve the screening process by investing in the infrastructure of high-performance computing. For each virtual drug screening project, our scientists will discuss with customers and then design a suitable and cost-effective screening solution.
References
- Neves B J.; et al. QSAR-based virtual screening: advances and applications in drug discovery. Frontiers in pharmacology. 2018, 9: 1275.
- Banegas-Luna A J.; et al. A review of ligand-based virtual screening web tools and screening algorithms in large molecular databases in the age of big data. Future medicinal chemistry. 2018, 10(22): 2641-2658.