De Novo Drug Design
De novo drug design is a computational technique wherein novel molecular structures with desired pharmacological properties are generated from the beginning or scratch. This technique is complementary to high-throughput screening and in silico virtual screening and is believed to contribute to the pharmaceutical development of novel drugs with desired properties at an extremely low cost and time-efficient manner. Creative Biostructure offers de novo design solutions to help you identify new leads or to produce novel molecular scaffolds and bioisosteric equivalent for already determined or undesired fragments.
According to specific project information, we adopt structure-based de novo drug design (SBDND) and ligand-based de novo drug design (LBDND). SBDND utilizes the three-dimensional (3D) structure of the target protein obtained from experimental structure determination approaches or target modeling. SBDND can be achieved by either a ligand growing or linking approach. The ligand growth approach involves docking a fragment at the binding site of the target protein and extending it by adding favorable functional groups. The linking approach is to dock multiple small fragments in the binding pocket of the target molecule and link them to form one complex molecule. In the absence of target 3D structure, the LBDND strategy can be used, which exploits structure-activity relationship (SAR) information about the existing active and inactive compounds.
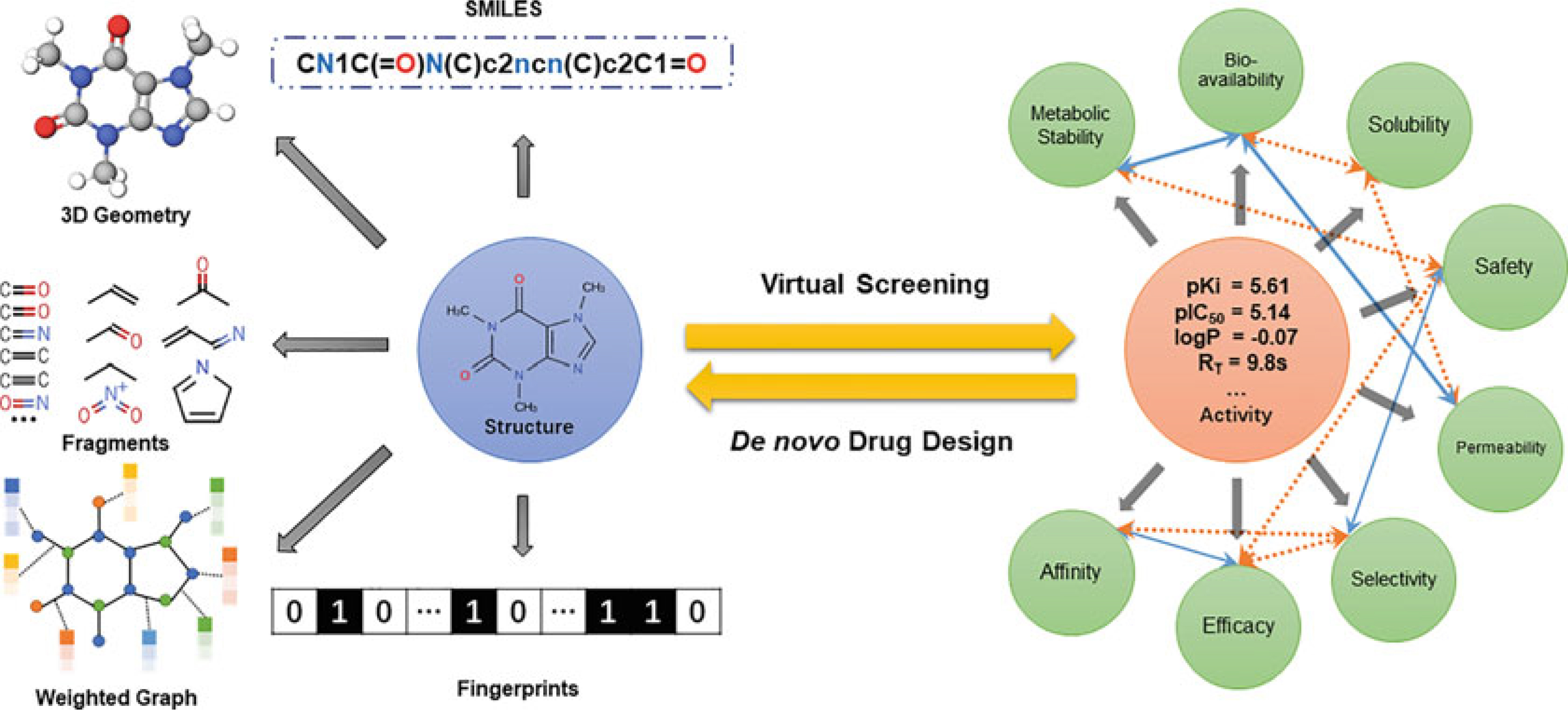 Figure 1. Schematic of the interplay of two approaches in computational drug discovery: virtual screening and de novo design. (Liu X.; et al. 2020)
Figure 1. Schematic of the interplay of two approaches in computational drug discovery: virtual screening and de novo design. (Liu X.; et al. 2020)
There are several computational methods for de novo design:
| Identifies a specific position of atom or fragment in binding sites. |
| Identifies a unique site location for placing fragments in the binding sites. |
| Connects the fragments at a particular position within binding sites. |
| Builds a library of fragments with desirable information. |
| Keep atoms or fragments at different places within binding sites of target and growing them by joining with other atoms/fragments with various coordination. |
| Connect the fragments in a random way. |
We follow three paradigms recognized by the de novo design program:
1. Structure construction of the candidate compounds (atom/fragment-based).
2. Evaluation of the molecule class, such as its fitness (based on 3D protein-ligand docking and scoring or ligand-based similarity measure).
3. Examining the search space or difficulty of optimization (based on depth-first/breadth-first search, evolutionary algorithms, exhaustive structure enumeration).
Capabilities and advantages of our De Novo Drug Design services:
- De novo drug design not only generates chemical starting points/hypotheses but also enables to secure intellectual property rights.
- We have established a computer-aided drug design (CADD) platform, which covers a variety of powerful computing software and tools.
- As a leading contract service provider in structural biology, we have extensive experience in target structure determination.
- We have rich expertise and experience in quantitative structure-activity relationship analysis based on 3D structures of molecules (3D-QSAR), which is an important and widely used method in LBDND.
- De novo design based on deep learning can accelerate the identification of new lead compounds.
- We guarantee the confidentiality of the information and data of each project.
Creative Biostructure's drug discovery team specializes in structure-based drug design and ligand-based drug design. Our scientists tailor professional and cost-effective solutions for each drug discovery-related project, control the key links of project execution, and provide professional result analysis.
References
- Schneider G, Fechner U. Computer-based de novo design of drug-like molecules. Nature Reviews Drug Discovery. 2005, 4(8): 649-663.
- Suryanarayanan V.; et al. De novo design of ligands using computational methods. Computational Drug Discovery and Design. Humana Press, New York, NY, 2018: 71-86.
- Schneider G, Clark D E. Automated de novo drug design: are we nearly there yet?. Angewandte Chemie International Edition. 2019, 58(32): 10792-10803.
- Liu X.; et al. Computational approaches for de novo drug design: past, present, and future. Artificial Neural Networks. Humana, New York, NY, 139-165.